
�й���ɫ����ѧ�� 2004,(04),534-538 DOI:10.19476/j.ysxb.1004.0609.2004.04.003
��������Ni75AlxV25-x�Ͻ����ڳ������̵�ԭ�ӳ߶ȼ����ģ��
������ҵ��ѧ���Ͽ�ѧ�빤��ѧԺ,������ҵ��ѧ���Ͽ�ѧ�빤��ѧԺ,������ҵ��ѧ���Ͽ�ѧ�빤��ѧԺ,������ҵ��ѧ���Ͽ�ѧ�빤��ѧԺ ����710072 ,����710072 ,����710072 ,����710072
ժ Ҫ��
�������ೡģ��,������ɢ�����ʽ������ɢ���̺ͷ�ƽ�������ܺ���,��������ԪNi75AlxV25-x�Ͻ��ԭ�Ӳ�������ģ�����ģ�ⷢ��:��������Ni75AlxV25-x�Ͻ�Ħ������ڦá�������,���������Ϊ�ȳɷ�����+ʧ�ȷֽ�;�á����ڦ������紦�Ǿ����κ�,���߾����γɷǻ�ѧ������������,֮����ѧ������������ת�䡣
�ؼ��ʣ�
��ͼ����ţ� TG111
����飺�����(1974),Ů,��ʿ.;
�ո����ڣ�2003-03-25
����������Ȼ��ѧ����������Ŀ(50071046);
Atomic-scale computer simulation for early precipitation process of Ni75AlxV75-x alloys with lower Al concentration
Abstract��
With the microscopic phase-field model, the atomic-scale computer simulation programs of the ternary Ni-based alloys were worked out based on the microscopic diffusion equation and nonequilibrium free energy. The results show that for Ni75AlxV25-x alloy with lower Al composition, �� ordered phase precipitates earlier than �á� ordered phase does by congruent ordering and spinodal decomposition mechanism, and thus produces a nonstoicheometric �� single ordered phase. Then, the nonstoicheometric �á� phase precipitates by a non-classical nucleation and growth mechanism at the APBS of �� phases.
Keyword��
early precipitation process; atomic-scale; computer simulation;
Received�� 2003-03-25
�Ͻ����ڳ�����ʱ��߶����뼶, �ռ�߶�����ʮ������ԭ��, ���γ�������ɢǿ����, ���е�ʵ���ֶ��о��Ͻ�������ڵĹ��ɺͻ��Ʒdz�����, ����ԭ�Ӳ���ļ����ģ���о����б�Ҫ��, ����ʾ��ʵ���ֶ��ɱ������Խ�ԡ� Pareige
������������Ԫ��ϵ����ɢ����, ͨ���Գ���������ԭ��ͼ�� ������ȵ�ģ��ͼ������, �Ե�������Ni-Al-V��ϵ���ڳ���������������(Ni3Al)������(Ni3V)����ʱ��������������м���ͬ�������ƽ������о���
1��Ԫ��ϵ���ೡ��ɢ����
��ģ��������Khachatuyran
ʽ�� L����(r-r��)Ϊ�뵥λʱ����1��������ԭ���ڸ��λ��r��r���ϵĽ��������йصij���, ��, ��=A, B��C; FΪ��ϵ����������, ����ƽ����������; TΪ�¶�; kBΪ��������������
ʽ(1)Ϊȷ������, ���������κ˹���, �������������ģ�������, ʹ֮��Ϊ�������, ��������Ҷ�任��, �õ�����Ҷ�ռ��е���Langevin����
ʽ�� kΪ��һ����Ԩ������ĵ���ʸ;
��ŷ��������ⷽ��, �õ���ͬʱ�̵�ԭ��ռλ����, �����õ�ԭ��ͼ�� ������ֲ��ȡ�
2ģ���������
ͼ1��ʾΪ��ƽ�������ۼ����Ni3Al-Ni3Vα��Ԫ��ͼ�� ��ͼ�ϴ�ʵ��Ϊƽ�������, ���ߡ�L12line��ΪL12������ʧ����, ���·���������L12�ṹ��������ʧ��; �㻮�ߡ�DO22line��ΪDO22������ʧ����, ���·���������DO22�ṹ��������ʧ�ȡ� 2������ʧ���߽����Ӧ��������Ϊ5.3%�� ���Ľ����ೡ�����»���Ϊ3������: �������������Ͻ�, �ɷַ�ΧΪ0.8%��5.0%; �����м��������Ͻ�, �ɷַ�ΧΪ5.0%��6.0%; �������������Ͻ�, �ɷַ�ΧΪ6.0%��1.7%�� ����ѡȡλ�ڢ�����Ni75Al3.2V21.8�Ͻ�Ϊ�о�����
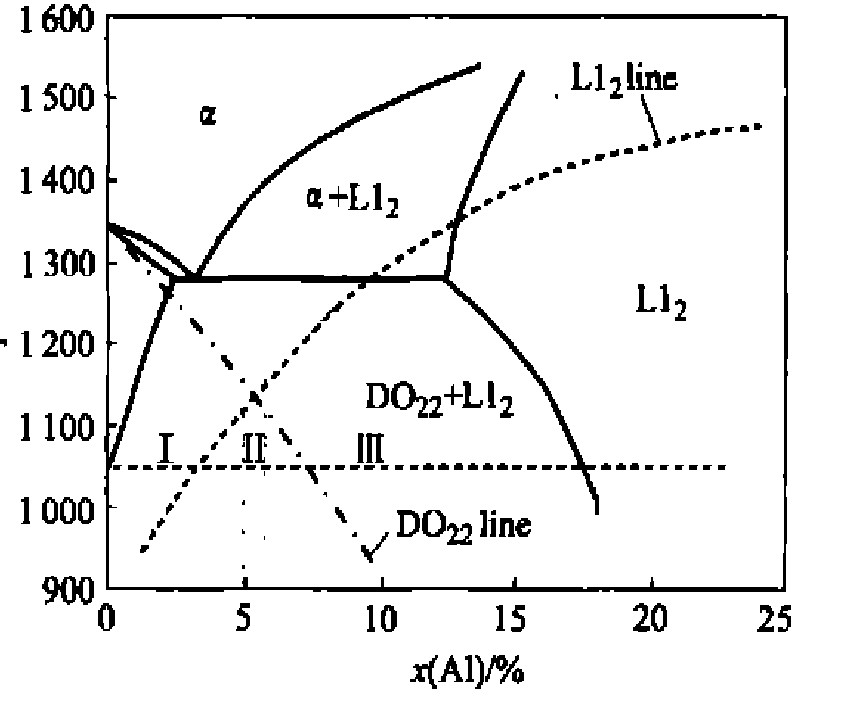
ͼ1 ��ƽ�������ۼ���� Ni-Al-Vα��ԪNi3Al-Ni3V��ͼ
Fig.1 Computed pseudobinary Ni3Al-Ni3V phase diagram with mean field model
ͼ2��ʾΪ��ͬʱ�䲽�ϸúϽ��ģ��ԭ��ͼ����ݻ�,ͼ3 ��ͼ4��ʾ�ֱ�Ϊ�úϽ�������������ྦྷ���ڲ��ijɷ�������ͳ���������ֲ���ʱ��ı仯��
ͼ2��ʾΪ������Ϊ3.2%ʱ, ��ͬʱ�䲽�ϺϽ��ģ��ԭ��ͼ���ݻ�����,ʱ�䲽��Ϊ0.000 2, �����Ϊ128��128, ��, ���ͷ�ԭ����ÿ������ϵ�ռλ����ֵ��ͬ, �Ӷ����ɲ�ͬ������ṹ�� ͼ2(a)��ʱ�䲽��Ϊ2 000, ��ʱ�Ͻ��Դ����������״̬�� ͼ2(b)��ʱ�䲽��Ϊ4 600, ������忪ʼ��������ת��, �����г���һЩDO22����ṹ�� ͼ2(c)��ʱ�䲽��Ϊ6 000, �Ͻ��л�����ȫ�γ�DO22�ṹ��������뼰��90����ת��, �����֮�䱻������(APBS)����, ��ԭ��������紦�ۼ��� ͼ2(d)��ʱ�䲽��Ϊ15 000, ����ЩAPBS����ʼ������һ��L12�ṹ����롣 ��ͼ2(e)��2(f)�пɹ۲쵽��������Ľ�һ���ֻ��� ������ļ��������µ���������κˡ�
ͼ3��, ��t=5 000ʱ, �ɷ�������ֲ���ƽ, û�����, ��ͬ��ʱ�䲽���ϵij���������Ѿ��ﵽ���ֵ, ��Ϊ���͵ĵȳɷ�������, �������γɵ��Ƿǻ�ѧ��������������, ���ɷֵ���ƽ��ֵ������������Ѵ�ƽ��ֵ�� ������, �ɷ������������Χ������̶Ƚ�С�����, ��������ƽ��ֵ, ��Ϊ��ѧ��������������, �Ҿ������Ե��м䰼�� ��ͷ������, ��������ѡ��������������紦�����������κ�, ��ԭ�Ӵ�������������ڲ���ɢ���¡� �������������������, �������С, ��ӳ���������խ, ���ڳɷ�������ֲ���ƽ, ˵����ԭ������ɢ�������в��ﵽƽ�⡣

ͼ2 ������Ϊ3.2%�ĺϽ���1 050 KʱЧʱ����ԭ��ռλ������ʱ����ݻ�Fig.2 Temporal evolution of occupation probabilities solute atoms at 1 050 K for alloy with 3.2%Al
(a)��t=2 000;(b)��t=4 600;(c)��t=6 000;(d)��t=15 000;(e)��t=20 000;(f)��t=100 000
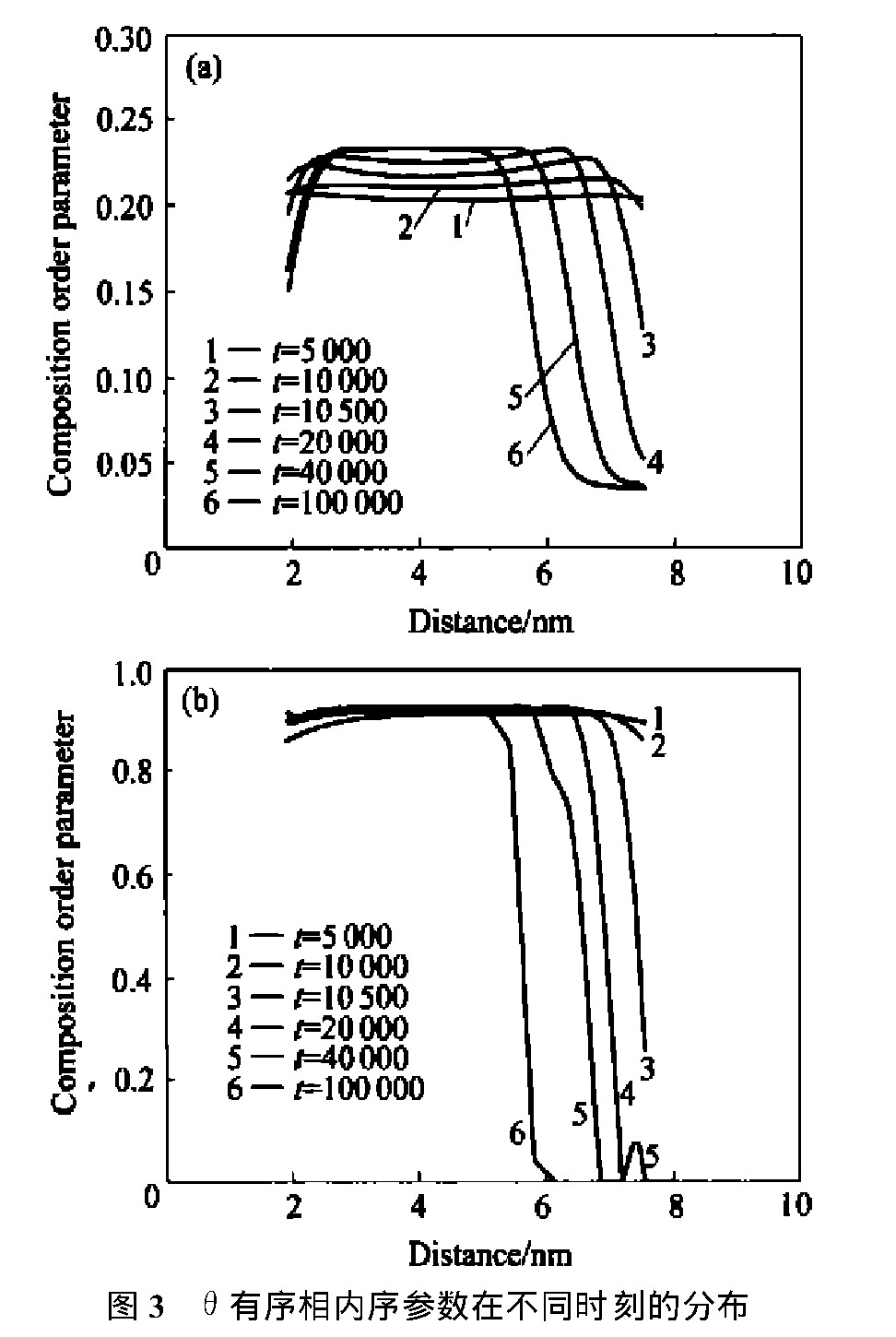
ͼ3 ����������������ڲ�ͬʱ�̵ķֲ�Fig.3 Order parameter profiles across��ordered phase for alloy with 3.2%Al at different times
(a)��Composition order parameter;(b)��Long-range order parameter
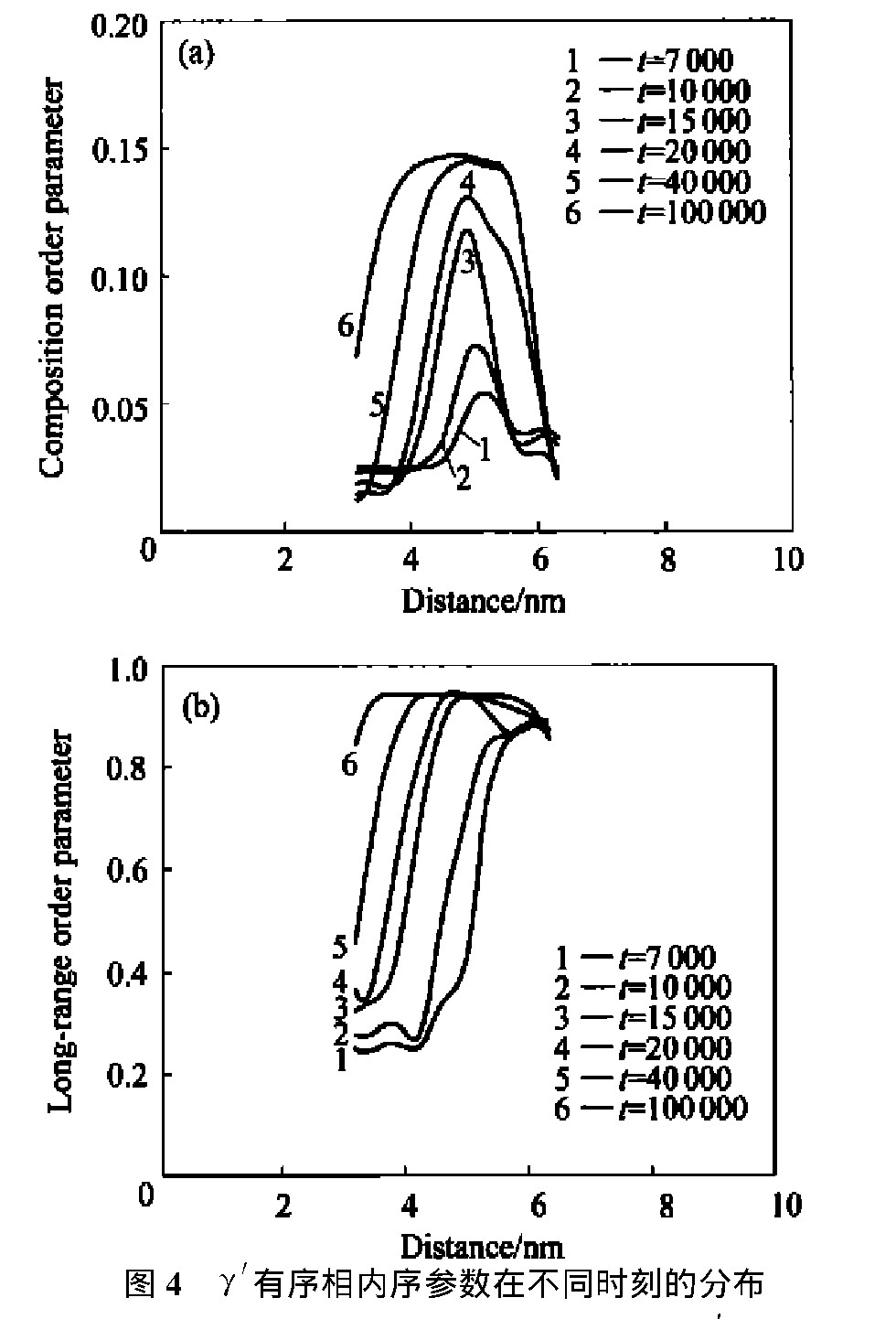
ͼ4 �á���������������ڲ�ͬʱ�̵ķֲ�Fig.4 Order parameter profiles across�á�ordered phase for alloy with 3.2%Al at different times
(a)��Composition order parameter;(b)��Long-range order parameter
��ͼ4�ɼ�, �������������ڳɷ�������ֲ�����Ǿ����κ˵��ص�: ���ijɷ�ֵ�ϵ�, ԶС��ƽ��ֵ; �ɷֲַ�������洦��һ���Ŀռ���չ�߶ȡ� ��ϳ��������, ��Ϊ���γɷǻ�ѧ�����������ࡣ ����ʱЧʱ����ӳ�, ���ijɷ�ֵ�ӽ�ƽ��, ת��Ϊ��ѧ�����������ࡣ Ni75Al3.2V21.8�Ͻ�ij���������ͼ5��ʾ��
3������ʵ��Ķ���
������Ϧ�������ݴ�ѧBendersky��
Zapolsky��
ͼ7��ʾΪPareige�������ؿ��巽����Ni78.3Al6.6V15.1�Ͻ���800 �����ʱЧ�����ڹ��̽����о�
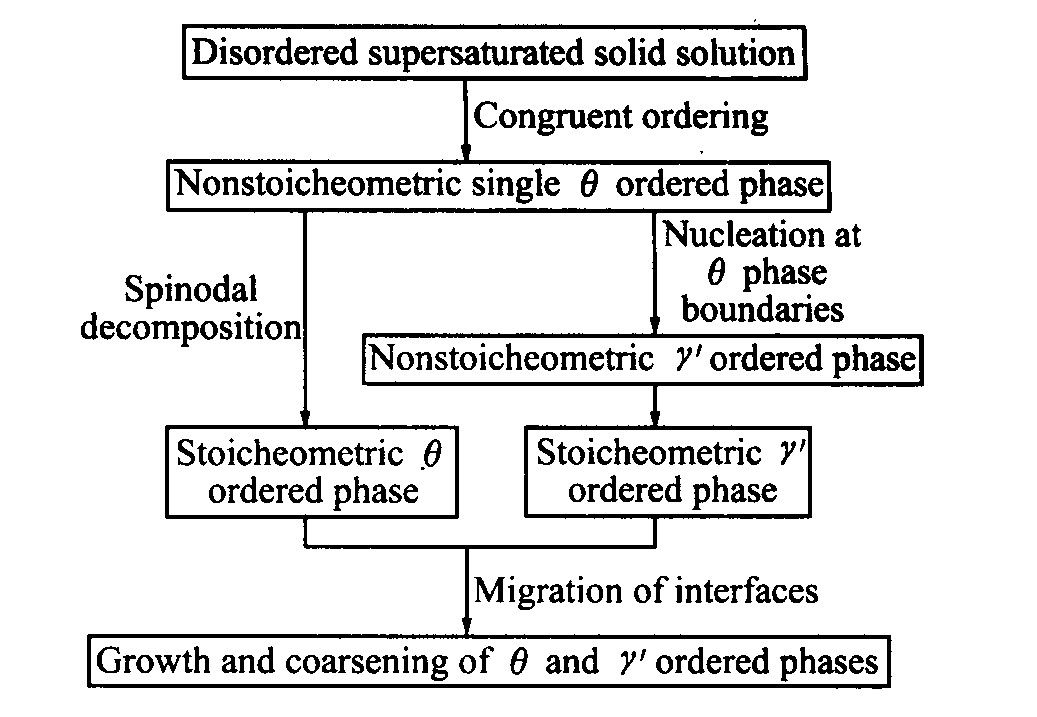
ͼ5 Ni75Al3.2V21.8�Ͻ�ij�������
Fig.5 Precipitate process of Ni75Al3.2V21.8 alloy
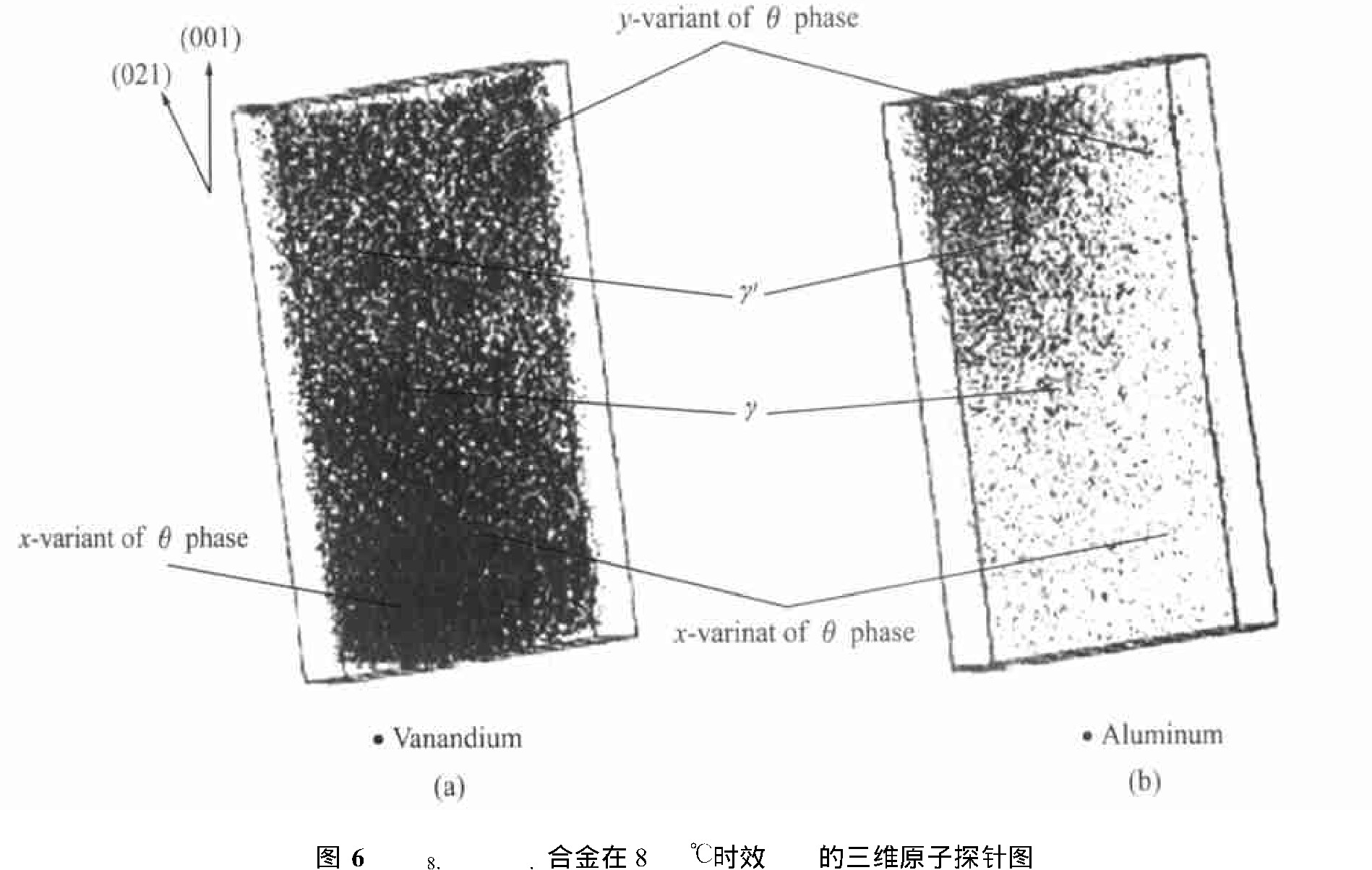
ͼ6 Ni78.5Al7V14.5�Ͻ���800��ʱЧ1 h����άԭ��̽��ͼFig.6 3D atomic probe maps for Ni78.5Al7V14.5alloy aged at 800��for 1 h
(a)����phase;(b)���á�phase
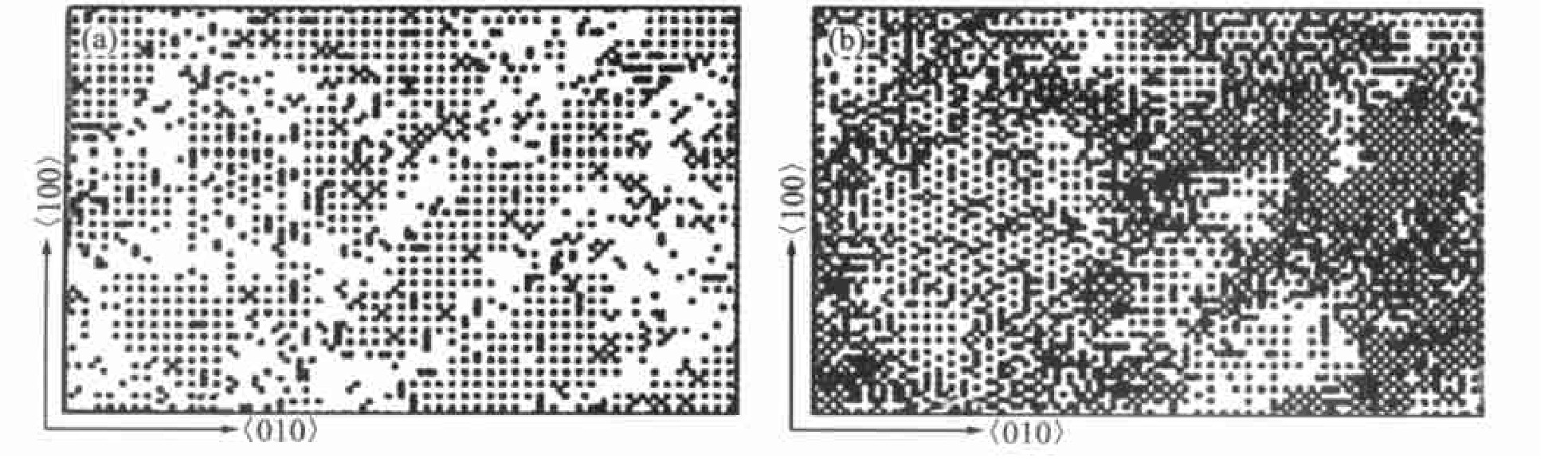
ͼ7 Ni78.3Al6.6V15.1�Ͻ���800��ʱЧʱ��ģ��ͼFig.7 3D elemen tmaps of Ni78.3Al6.6V15.1alloy aged at 800��with Monte-Carlo simulation
(a)��L12long ordered zones;(b)��DO22long ordered zones
4����
1) ��������Ni75AlxV25-x�Ͻ��������������ѧ��������2���������������, �ֱ���DO22�ṹ�����������L12�ṹ������������, 2���������α��Ԫ��ϵ��
2) 2�����������������Ϊ: ����������������������������
3) 2��������ij�������Ϊ: ԭ�ӵĵȳɷ��������γ�DO22�ṹ�ĵ����������, �����֮�䱻������(APBS)����, �����ʧ�ȷֽ�, ��ԭ�ӿ�ʼ��������������Ͼۼ�, ͬʱ��ԭ�Ӵ�APBS�ϼ��١� ������ԭ�������Ͼۼ��̶ȵ�����, ���������ʼ�κ˺ͳ��� ��������ij�������Ϊ�ȳɷ�����+ʧ�ȷֽ�, ������ij�������Ϊ����Ǿ����κˡ�
4) ���߾����γɷǻ�ѧ������������, Ȼ����ѧ������������ת�䡣
�����
[3] ��KhachatuyranAG.TheoryofStructuralTransformationsinSolids[M].NewYork:Wiley,1983.139.
[8] ��BenderskyLA.Solid SolidPhaseTransfomations[M].Warrendale,PA:TMS,1994.899902.
