
���±�ţ�1004-0609(2007)07-1160-06
LaNi4.5Al0.5����Ͻ��������ܶȷ����о�
�� ��1��������1���౾��1��������1���� ��2���Ŷ���1
(1. ����ʦ��ѧԺ �������ӹ���ѧԺ������ 464000��
2. �Ĵ���ѧ ԭ������������о������ɶ� 610065)
ժ Ҫ��
�����ܶȷ������ۣ�����ȫ��������ƽ�沨����(FLAPW)���о�LaNi4.5Al0.5����Ͻ�������-LaNi4.5Al0.5H0.5�ͦ�-LaNi4.5Al0.5H1.0��Hԭ�ӵ�ռλ��̬�ܶȺ͵����ܶȣ�������Hԭ�ӵļ���Թ�������ӽṹ���ȶ��Ե�Ӱ�졣����������������Ƕȼ���õ���-LaNi4.5Al0.5H0.5��Hԭ�������ռ�ݿ���Al��6mλ����-LaNi4.5Al0.5H1.0�е�����Hԭ�������ռ��6m��4h*λ������Hԭ�ӵ����ӣ�������Ҫ����c�᷽�����ͣ�Al��Ni, H ֮���������ǺϽ�������屣���ȶ�����Ҫ���أ�̬�ܶ�ͼ�е����������̬�ܶ�Խ�������Խ�ȶ������EF���ڴ�϶�ĵײ�������ϵ���ȶ��������������е�ʵ�����dz�һ�¡�
�ؼ��ʣ�
LaNi4.5Al0.5�Ͻ����ܶȷ���������������ƽ�沨����(FLAPW)����������
��ͼ����ţ�TG 139.7���� ���ױ�ʶ�룺A
Density functional theory study on solid solution phase
of LaNi4.5Al0.5 hydrogen storage alloys
CHEN Dong1, ZHOU Li-hai1, YU Ben-hai1, WANG Chun-lei1, GAO Tao2, ZHANG Dong-ling1
(1. School of Physics and Electronic Engineering, Xinyang Normal University, Xinyang 464000, China;
2. Institute of Atomic and Molecular Physics, Sichuan University, Chengdu 610065, China)
Abstract: Based on the density functional theory (DFT) and full-potential linearized augmented plane wave (FLAPW) method, the hydrogen occupied sites, electron densities and densities of states were analyzed for the solid solution phase ��-LaNi4.5Al0.5H0.5 and ��-LaNi4.5Al0.5H1.0. The hydrogen atom in ��-LaNi4.5Al0.5H0.5 is found to prefer the 6m position near aluminum atom, the two hydrogen atoms in ��-LaNi4.5Al0.5H0.5H1.0 are most likely to take the 6m and 4h* sites by total energy minimization calculation. The lattice expansion is mainly along the c axis. The interaction between aluminum and nickel, hydrogen plays a dominant role in the stability of LaNi4.5Al0.5Hx solid solution phase. The smaller the shift of EF towards higher energy region, the more stable the compounds will be. The calculated results are compared with the existent experimental data and discussed in light of previous works.
Key words: LaNi4.5Al0.5 alloys; density functional theory; full-potential linearized augmented plane wave (FLAPW) method; solid solution phase
LaNi5�Ͻ���ϡ��ϵ����Ͻ�ĵ��ʹ����������д����������ƽ��ѹ�����С��������ٶȿ���ŵ�[1-2]��Ϊ������ʵ�ʹ�������Ҫ�������ý���Mȡ������Ni�γɺϽ�LaNi5-yMy(M=Fe��Co��Mn��Al��)������Al���۴Ӽ۸��ȶ��Ժ��������ȽǶȶ�������Խ�ԡ���Al��������ʱ���Ͻ�Ĵ��������ٲ��࣬�����ȶ��Ժ�����ѧ���ʶ��ܵõ����Ը��ơ�
�����������Ƕ�La-Ni-Al�Ͻ����������������о�����Ϊ�ڹ������У�Hԭ��ֻռ�ݲ��ָ�λ����Ͻ�ṹ��Ȼ���ֲ��䡣Ȼ����Ŀǰ���ںϽ��������Hԭ�ӵ�ռλ������ӽṹ��ʵ���о���Ȼ���٣����˽����ںϽ��е�ռλ�Լ��ɴ�����ĺϽ���ӽṹ�ı仯�����Ը��õ��о�����ṹ�ĸ����������ͺͶԺϽ�����ѧ���ʵ�Ӱ�죬�ԺϽ���Ż������ָ�����á�
�����������ܶȷ������ۣ�����ȫ��������ƽ�沨(Full-potential linearized augmented plane wave, FLAPW)��������ԭ��ˮƽ�ϼ������LaNi4.5Al0.5Hx�ľ���ṹ���״η���Hԭ�����п���ռ�ݵĸ�λ��������Al��H�Ծ��弸�νṹ�͵��ӽṹ��Ӱ�졣
1 ����ģ�ͼ����۷���
LaNi5����CaCu5�;���ṹ������Laռ1a(0,0,0)��Niռ��2c(0.3333,0.6667,0)��3g(0.5,0, 0.5)Wyckoff��λ������������ϵ���ռ�ȺΪP6/mmm��Ϊ�˽��LaNi4.5Al0.5��Al�����Ƿ��������⣬��ģ���й�����˫����ģ�ͣ���z��������LaNi5������Ȼ��ʹ��Alԭ��ȡ��һ��Niԭ�ӡ�Jensen��[3]ͨ��X���߷�ĩ���䷨������Ϊ������LaNi5-xAlx���ԣ���x��1.3ʱ���Ͻ��Ա���CaCu5�ṹ��Szajek[4]��LaNi4Al���о���Ϊ��Alԭ������ռ��3g��λ�� Percheron[5-6]��ΪAlԭ��ֻ��ռ��3g��λ��Zhang����[7]��Ϊ��Ԫ�Ͻ�LaNi4.5Al0.5��Alԭ��֮�䲻�����ڡ���˱������߹�����Alԭ�����3g��λ(0.5a, 0.5a, 0.75c)����Niԭ�ӵ�˫����ģ�ͣ����к���2��Laԭ�ӡ�9��Niԭ�ӡ�1��Alԭ�ӣ��ù�ʽ��ʾ������Ϊ2La+9Ni+ Al=2( LaNi4.5Al0.5)����ṹʾ��ͼ��ͼ1��ʾ��
���������ȫ��������ƽ�沨�������÷����Ǽ��㾧����ӽṹ�ȷ�ķ���֮һ���ڼ��������ȴ��ܶȷ������۳�����������Ķ����Schr?dinger���̻�Ϊ������Schr?dinger����(Kohn-Sham����)��Ȼ��ͨ������������ƽ�沨�������Ե�����Kohn-Sham���̽��м��㣬ͬʱ��������ݶȽ�������(GGA)�������ӵĽ����ܱ�ʾΪ�����ܶȼ����ݶȵĺ��������ڸ����в�ͬ�ļ�����������ʹ��PBE96[8]����LaNi5�ļ���������÷�����ʵ������Ϊ�ӽ�[9]��������ƽ�沨�����л�������ѡȡ�Ͷ�Kohn-Sham������Ǣ�����ʱ����ģ�͵Ľ��������á�Muffin-Tin��ģ�͡�

ͼ1 LaNi4.5Al0.5��˫�����ṹ
Fig.1 Structure of double unit cell of LaNi4.5Al0.5
���������WIEN2k���������������Ѱ����ڸó�����[10]��
2 ������������
��ģ�������м��㶼�����ܶȷ������ۿ���½��еġ�������La��Ni��Al��H��Muffin-tin�뾶�ֱ�ȡΪ1.32��1.23��1.15��0.37 nm�����г�ʼ��ʱ������PBE96�����������ӵĽ�������ܣ�ѡȡ���� -81.6 eV�ֿ��ڲ���Ӻͼ۵��ӣ���ԭ�ӵĵ�����̬�ֱ�Ϊ��La 5s25p65d16s2��Ni 3p63d84s2��Al 3s23p1��H 1s1������Monkhorst-Pack�������ڲ���Լ����Ԩ����ȡ300��k�㣬ƽ�沨�ض�����ȡ300 eV��������г������չ��ָ��ȡΪ12.0�����нṹ�Ż�ʱ�����������������淨��1) ����c/a���䣬�仯���V��2) �������V���䣬�仯c/a��ֵ����������ֱ����У�ֱ��ǰ�����μ�����֮��С��1%�������������Ǣ�������������������ȿ���Ϊ������0.001 36 eV�����������ţ����ѧ������ÿ��ԭ�ӵ�ƽ��λ�ý��г����Ż����ڼ����п��������ЧӦ��
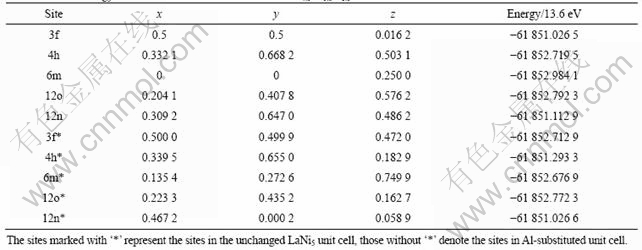
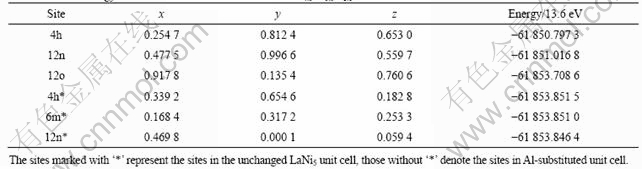
���ڻ�ѧ�����ܴ��������ı仯�����Hԭ�ӵļ��뽫ֱ��Ӱ�쵽�Ͻ�ľ�������͵������á�����Westlake[11]����ģ�͵Ĺ���ԭ������ĸ����������У�Hԭ����Ҫռ��������Ͱ������϶�����������뾶����0.04nm�Ŀ�϶��LaNi5�����й�����3f��4h��6m��12n��12o��5����λ����϶�뾶�ɴ���С��˳��Ϊ��6m��12n��12o��4h��3f[12]����10�ֲ�ͬ��λ�и�����һ��Hԭ�ӵ�˫�����м��㣬���Hԭ��ռ�ݺθ�λ������������ͣ����������оݣ���Ϊ��λ�þ���Hԭ�������ռ�ݵ�λ�á�Ϊ�˵õ����Ž�����Ծ������нṹ�Ż����������ԥ��������1�ͱ�2���С�
��1�ͱ�2�в�����*���ŵĿ�϶λ�ô�������Alԭ��һ��ĸ�λ���ɱ����Կ�����Hԭ��ȡ��6m��λ�������������ͣ�Hԭ�������ռ��6mλ����������Du��[13]�����������䷨��LaNi5-xAlx���о����һ�¡�����Ono��[14]����Hԭ�ӽ���˳��ļ����֪��6m��λ�����������ڻ�ƽ��Ŀ�϶λ���ҿ�϶�뾶�ϴ��ǵ�Ono�ȵ����������λ�Ĵ�С��ϵ���Եڶ���Hԭ��ֻ����6������ܵ�ģ�ͣ����㷢��4h*��λ��Hԭ�������ռ�ݵ�λ�á��ɴ˿ɼ���Hԭ�Ӳ����ǰ��տ�϶��С˳�����Ͻ�ģ���ܿ����������ȼ����Hԭ��ʹ��λ�������䣬Ӱ���˵ڶ���Hԭ�ӽ���ĸ�λ��
LaNi4.5Al0.5�ľ�����Ϊ��a=0.504 3 nm��c=��0.403 0 nm[5]��LaNi4.5Al0.5H0.5�ľ�����Ϊ��a��0.504 3 nm��c��0.404 2 nm��LaNi4.5Al0.5H1.0�ľ�����Ϊ��a=0.505 5 nm��c=0.410 8 nm�����Կ���������Hԭ�ӵļ��룬������Ҫ��c�᷽�����͡�
2.2.1 LaNi4.5Al0.5H0.5��̬�ܶȷ���
ͼ2��ʾ��La�ķֲ�̬�ܶȡ�ͼ3��ʾ��LaNi4.5Al0.5H0.5��La��Ni��Al���ܲ�̬�ܶ�ͼ��ͼ����EF��-9.956 6 eVΪ��㣬��LaNi4.5Al0.5��EF����-0.165 9 eV[15]����LaNi5������0.069 7 eV[9]������Ҫ��Hԭ�Ӻ�Alԭ�ӵĹ��ף��Ҷ���s����Ĺ��״���p����ġ������ܼ�����n(EF)Ϊ8.89��ͨ��![]() [16]����ɵã�LaNi4.5Al0.5H0.5�ĵ��ӱ���ϵ��Ϊ20.97 mJ/mol������kBΪ��������������
[16]����ɵã�LaNi4.5Al0.5H0.5�ĵ��ӱ���ϵ��Ϊ20.97 mJ/mol������kBΪ��������������
��Ϊ�ڼ����в���˫�����ṹ�������ܲ�̬�ܶȺ�ԭ�ӵķֲ�̬�ܶȶ��ǵ�����2�����Ա�ͼ3��ͼ4��֪��Niԭ�ӵĵ���ռ���˵����ľ���Ȩ�أ�
��1 LaNi4.5Al0.5H0.5������Hԭ�ӵ�λ�ü�����������
Table 1 Total energy and interstitial sites in unit cell of LaNi4.5Al0.5H0.5
��2 LaNi4.5Al0.5H1.0������Hԭ�ӵ�λ�ü�����������
Table 2 Total energy and interstitial sites in unit cell of LaNi4.5Al0.5H1.0
�����ܼ�EF����̬�ܶ���Ҫ��Niԭ�ӵ�3d���ṩ����La��̬�ܶ���Ҫ�����ڼ۴�3.7��-16.0 eV������La��f���������˼۴�����Ҫ���֡��ܲ�̬�ܶ���Ҫ����Ni��3d��La��4f�����ɵģ���-7.1~-6.5 eV�������ҽ���һ����϶��
�ܲ�̬�ܶ�ͼ��-9~-5 eV����̬�ܶ���Ҫ��H-s��Al-s��Ni-s,p,d����Ĺ��ס��ڵ��������ڣ�Ni

ͼ2 La�ķֲ�̬�ܶ�
Fig.2 Partial DOS of La

ͼ3 La��Ni��Al�Լ�LaNi4.5Al0.5H0.5���ܲ�̬�ܶ�
Fig.3 Total DOS of La, Ni, Al and LaNi4.5Al0.5H0.5
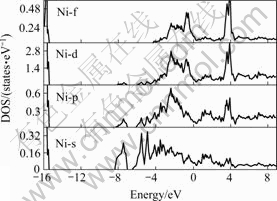
ͼ4 Ni�ķֲ�̬�ܶ�
Fig.4 Partial DOS of Ni
ԭ�Ӷ��ܲ�̬�ܶȵĹ������Alԭ�Ӵ�֮��Hԭ�ӵĹ�����С��Ҳ���ɺ��ԡ���ͼ3~5��֪��Ni��H֮������������ǿ�ģ�Al��H�������ô�֮��H��La֮����������С������Ȼ��H��Ni��Al֮����������LaNi4.5Al0.5H0.5�Ͻ𱣳��ȶ�����Ҫ���ء�

ͼ5 LaNi4.5Al0.5H0.5�Ͻ���Al��H�ķֲ�̬�ܶ�
Fig.5 Partial DOS of Al and H in LaNi4.5Al0.5H0.5 alloy
2.2.2 LaNi4.5Al0.5H1.0��̬�ܶȷ���
��ͼ6�ɼ���LaNi4.5Al0.5H1.0��Hԭ�ӵ�s��p��������LaNi4.5Al0.5H0.5��Hԭ��s��p�����Ľ�2������������Hԭ�����ӵ�������Ե�ʡ������ܼ�����n(EF)=8.91��LaNi4.5Al0.5H1.0�ĵ��ӱ���ϵ��Ϊ21.02 mJ/mol������Hԭ�ӵļ��룬����������������-10.122 5��-9.956 6���ߵ�-9.875 0 eV����EF����̬�ܶȴ�8.86��8.89���ӵ�8.91 states/eV������H��Al�Ĺ��ס�
��LaNi4.5Al0.5H1.0��La-4f�����Ni-3d��������ܲ�̬�ܶȵ���Ҫ�����ߡ�ͼ7��LaNi4.5Al0.5H1.0���ܲ�̬�ܶ���LaNi4.5Al0.5H0.5����ȱ仯Ҳ�����ԣ�
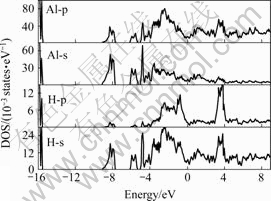
ͼ6 LaNi4.5Al0.5H1.0�Ͻ���Al��H�ķֲ�̬�ܶ�
Fig.6 Partial DOS of Al and H in LaNi4.5Al0.5H1.0 alloy
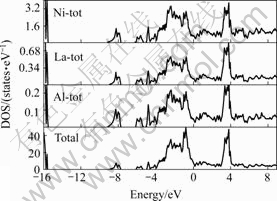
ͼ7 La��Ni��,Al�Լ�LaNi4.5Al0.5H1.0���ܲ�̬�ܶ�
Fig.7 Total DOS of La, Ni, Al and LaNi4.5Al0.5H1.0
�����������Hԭ�Ӻ������ٺ�H���ܲ�̬�ܶȵĹ���С��Ե�ʡ�La��̬�ܶ���Ҫ������3.5 eV������϶�Ŀ��ȱȦ�-LaNi4.5Al0.5H0.5Ҫ�������ǰ�������ɵġ�
��LaNi5���ܲ�̬�ܶ����[15]��LaNi4.5Al0.5H0.5��LaNi4.5Al0.5H1.0(ͼ3��7)���ܲ�̬�ܶ��ڵ������������Ӵ�������-6 eV������̬�ܶ�����Al��H��Ni��H֮������������ϵ�ģ�-5 eV������̬�ܶ�������Ni��Al������������ġ����ھ������ͣ�La��Ni����������������������Hԭ�ӵļ��룬�����ܼ�����̬�ܶ���������ϵ���ȶ��Խ��͡�ʵ��õ�Ӱ����ϵ�ȶ��Ե��������طֱ�Ϊ��1) ����������̬�ܶ�Խ����ϵԽ�ȶ���2) ���EF����α��϶�ĵײ�������ϵ���ȶ���
�˽�����ܶȵĿռ�ֲ����Է��������е�ԭ�ӳɼ�״�����о��������Զ��Ƿdz���Ҫ�ġ�ͨ����ɿռ��ܶȷֲ��Ϳ��Եó�������ԭ�ӵijɼ�״����
ͼ8��9��ʾ�ֱ�ΪLaNi4.5Al0.5H0.5(![]() )���LaNi4.5Al0.5H1.0(
)���LaNi4.5Al0.5H1.0(![]() )��ĵ����ܶ�ͼ��ͼ��û�г��־���ļ���ɣ�˵������ԭ����Hԭ��֮����ڵ��ǽ���������ͼ8�ɼ���Hԭ�ӵĵ����ܶ���Ҫ����Ni��H������ɢ��Al��H֮�������ñ�Ni��H֮����������������La��H֮��������ǿ��Alԭ����Ȼֻ�ǽӽ����棬������Niԭ��֮���Դ���һ��������ã���ܿ����ǺϽۻ�������ǿ��ԭ��Laԭ����Hԭ�ӵĵ����Ƽ���û�з�����������˵��H��La�����⻯����γ�Ԫ��[16]֮��Ļ�ѧ�����������뵥��Ԫ��La��H֮�����γɺ�ǿ�Ļ�ѧ������ȫ��ͬ�ġ�
)��ĵ����ܶ�ͼ��ͼ��û�г��־���ļ���ɣ�˵������ԭ����Hԭ��֮����ڵ��ǽ���������ͼ8�ɼ���Hԭ�ӵĵ����ܶ���Ҫ����Ni��H������ɢ��Al��H֮�������ñ�Ni��H֮����������������La��H֮��������ǿ��Alԭ����Ȼֻ�ǽӽ����棬������Niԭ��֮���Դ���һ��������ã���ܿ����ǺϽۻ�������ǿ��ԭ��Laԭ����Hԭ�ӵĵ����Ƽ���û�з�����������˵��H��La�����⻯����γ�Ԫ��[16]֮��Ļ�ѧ�����������뵥��Ԫ��La��H֮�����γɺ�ǿ�Ļ�ѧ������ȫ��ͬ�ġ�
�ڹ������п�����ΪH��Ni���������ǺϽ�
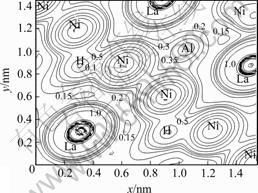
ͼ8 LaNi4.5Al0.5H0.5 (![]() )��ĵ����ܶ�
)��ĵ����ܶ�
Fig.8 Charge density in (![]() ) plane of LaNi4.5Al0.5H0.5 (103eV/nm3)
) plane of LaNi4.5Al0.5H0.5 (103eV/nm3)

ͼ9 LaNi4.5Al0.5H 1.0(![]() )��ĵ����ܶ�
)��ĵ����ܶ�
Fig.9 Charge density in (![]() ) plane of LaNi4.5Al0.5H1.0 (103eV/nm3)
) plane of LaNi4.5Al0.5H1.0 (103eV/nm3)
����ͷ������Ҫ������Niԭ�Ӻ�Laԭ��֮����������LaNi5������������������LaNi4.5Al0.5H1.0��c����0.403 0 nm[18]���͵�0.410 9 nm������1.96%����a������С��LaNi5H3��c������(5.12%)[19-20]���ɼ�Alԭ�ӵļ�����Ч������˺Ͻ�Ŀ��ۻ�����������ѭ��������
3 ����
1) �����ܶȷ������ۺ�ȫ��������ƽ�沨�������о���-LaNi4.5Al0.5Hx (x=0.5, 1.0)�ĵ��ӽṹ��Hԭ��ռλ��ͨ�������ֹ������в��ȼ۸�λ�����������С��������Ϊ����һ��Hԭ��������ռ��6m��λ���ڶ���Hԭ�������ռ��4h*��λ���ȼ����Hԭ��������䣬Ӱ���˺���Hԭ��ռ�ݵ�λ�á�
2) Ӱ����ϵ�ȶ��Ե���������Ϊ��һ�ǵ���������̬�ܶ�Խ����ϵԽ�ȶ����������EF����α��϶�ĵײ�������ϵ���ȶ���
3) ����Hԭ�ӵļ��룬������Ҫ����c�᷽�����ͣ������������ǿ��H��Ni֮�������ý�ǿ��H��Al֮����������Խ�������H��La֮�������������Alԭ�ӵļ�����Ч����˺Ͻ�Ŀ��ۻ�������ѭ��������
REFERENCES
[1] Nakamura H, Nguyen-Manh D, Pettifor D G. Electronic structure and energetics of LaNi5, ��-La2Ni10H and ��-La2Ni10H14[J]. J Alloys Comp, 1998, 281: 81-91.
[2] Kisi E H, Wu E, Kemali M. In-situ neutron powder diffraction study of annealing activated LaNi5[J]. J Alloys Comp, 2002, 330/332: 202-207.
[3] Jensen J O, Bjerrum N J. Systematic B-metal substitution in CaNi5[J]. J Alloys Comp, 1999, 293/295: 185-189.
[4] Szajek A, Jurezyk M, Rajewski W. The electronic and electrochemical properties of the LaNi5, LaNi4Al and LaNi3CoAl systems[J]. J Alloys Comp, 2000, 307: 290-296.
[5] Percheron-Gu��gan A, Lartigue C, Achard J C. Neutron and X-ray diffraction profile analyses and structure of LaNi5, LaNi5-xAlx and LaNi5-xMnx intermetallics and their hydrides(deuterides)[J]. J Less-Common Met, 1980, 74: 1-12.
[6] Lartigue C, Percheron-Gu��gan A, Achard J C. Hydrogen (deuterium) ordering in the ��-LaNi5Dx (x��5) phases: a neutron diffraction study[J]. J Less-Common Met, 1980, 75: 23-29.
[7] Zhang R J, Wang Y M, Lu M Q, Xu D S, Yang K. First-principles study on the crystal, electronic structure and stability of LaNi5-xAlx (x=0, 0.25, 0.5, 0.75 and 1)[J]. Acta Materialia, 2005, 53: 3445-3452.
[8] Perdew J P, Burke K, Ernzerhof M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77: 3865-3868.
[9] ���»�, �� ��, ������, ��Ծѫ, �¡���, ���ǽ�. LaNi5�����������ṹ��ȫ���Ӽ���[J]. ԭ�����������ѧ��, 2004, 21: 366-372.
QI Xin-hua, GAO Tao, ZHU Zheng-he, LI Yue-xun, CHEN Bo, FAN Zhi-jian. Full-electronic calculations on the equilibrium structure and energy of LaNi5 crystal[J]. J At Mol Phys, 2004, 21: 366-372.
[10] Schwarz K, Blaha P. Solid-state calculation using WIEN2k[J]. Computational Materials Science, 2003, 28: 259-273.
[11] Westlake D G. A geometric model for the stoichiometry and interstitial site occupancy in hydrides(deuterides) of LaNi5, LaNi4Al and LaNi4Mn[J]. J Less-Common Met, 1983, 91: 275-292.
[12] Soubeyroux J L, Percheron-Gu��gan A, Achard J C. Localization of hydrogen (deuterium) in �CLaNi5Hx (x=0.1 and 0.4)[J]. J Less-Common Met, 1987, 129: 181-186.
[13] Du H L, Zhang W Y, Wang C S, Wang J Z, Han J Z, Yang Y C, Chen B, Xie C M, Sun K, Zhang B S. Neutron powder diffraction study on the structures of LaNi5-xAlxDy compounds[J]. Solid State Communications, 2003, 128: 157-161.
[14] Ono S, Nomura K, Akiba K. Phase transformations of the LaNi5-H2 system[J]. J Less-Common Met, 1985, 113: 113-117.
[15] �� ��, ���»�, �� ��. Al�Ͻ�LaNi5-xAlx�ĽṹӰ��[J]. �й���ɫ����ѧ��, 2005, 15: 1092-1099.
GAO Tao, QI Xin-hua, CHEN Bo. Alloying effects on electronic structures of LaNi5-xAlx[J]. The Chinese Journal of Nonferrous Metals, 2005, 15: 1092-1099.
[16] Hector L G Jr, Herbst J F, Capehart T W. Electronic structure calculations for LaNi5 and LaNi5H7: energetics and elastic properties[J]. J Alloys Comp, 2003, 353: 74-85.
[17] Van Vucht J H N, Kuipers F A, Bruning H C M, et al. Reversible room-temperature absorptions large quamtities of hydrogen by intermetallic compounds[J]. Philips Res Repts, 1970, 25: 133-140.
[18] �� ��, ������. LaNi5-xAlx����Ͻ���о�[J]. ϡ��, 1996, 22: 11-15.
KANG Long, LUO Yong-chun. Research on LaNi5-xAlx hydrogen storage alloys[J]. Rare Earth, 1996, 22: 11-15.
[19] ���»�, �� ��, �� ��. ��-LaNi5H0.5���-LaNi5H3��ȫ�����Ż�����[J]. ����ѧ��, 2005, 41(9): 910-916.
QI Xin-hua, GAO Tao, CHEN Bo. Full-electron calculation of ��-LaNi5H0.5 and ��-LaNi5H3[J]. Acta Metallurgical Sinica, 2005, 41(9): 910-916.
[20] �� ��, ������. LaNi5��������ľ���ṹ����[J]. ϡ�н��������빤��, 2004, 33: 239-241.
WANG Hong, LIU Zu-yan. Crystal structure analysis on maximum hydrogen capacity of LaNi5[J]. Rare Metal Materials and Engineering, 2004, 33: 239-241.
(�༭��������)
������Ŀ��������Ȼ��ѧ���й����������о�Ժ���ϻ���(10276027)
�ո����ڣ�2006-09-20�������ڣ�2007-03-12
ͨѶ���ߣ��� �����绰��13253854009; E-mail: chchendong2010@163.com
[15] �� ��, ���»�, �� ��. Al�Ͻ�LaNi5-xAlx�ĽṹӰ��[J]. �й���ɫ����ѧ��, 2005, 15: 1092-1099.
[18] �� ��, ������. LaNi5-xAlx����Ͻ���о�[J]. ϡ��, 1996, 22: 11-15.
[19] ���»�, �� ��, �� ��. ��-LaNi5H0.5���-LaNi5H3��ȫ�����Ż�����[J]. ����ѧ��, 2005, 41(9): 910-916.
[20] �� ��, ������. LaNi5��������ľ���ṹ����[J]. ϡ�н��������빤��, 2004, 33: 239-241.
