

Particle swarm optimization computer simulation of Ni clusters
ZHOU Ji-cheng(�ܼ̳�)1, 2, LI Wen-juan(���ľ�)2, ZHU Jin-bo(���)2
1. School of Electronic and Information Engineering, Central South University of Forestry and Technology,
Changsha 410004, China;
2. School of Physics and Electronics, Central South University, Changsha 410083, China
Received 7 June 2007; accepted 12 September 2007
Abstract:
The stable structures and energies of Ni clusters were investigated using particle swarm optimization(PSO) combined with simulated annealing(SA). Sutton-Chen many-body potential was used in describing the interatomic interactions. The simulation results indicate that the structures of Ni clusters are icosahedral-like and binding energy per atom tends to approach that of bulk materials when the atoms number increases. The stability of Ni clusters depends not only on size but also on symmetrical characterization. The structure stability of Nin clusters increases with the increase of total atom number n. It is also found that there exists direct correlation between stability and geometrical structures of the clusters, and relatively higher symmetry clusters are more stable. From the results of the second difference in the binding energy, the clusters at n=3 is more stable than others, and the magic numbers effect is also found.
Key words:
particle swarm optimization; Ni cluster; Sutton-Chen potential; magic numbers effect;
1 Introduction
Clusters of atoms are intermediate state between small molecules and solids and exhibit a rich variety of physical and chemical properties[1]. Small clusters have become one of the most exciting research areas in recent years[2-3]. This is not only because they can act as models for understanding localized effects in solids, but also they can serve as building blocks for developing new materials with uncommon properties for technological applications[4-5]. Among them, Ni nanoclusters are a fundamental part of recently synthesized nanocrystalline materials[6-7]. The geometrical structure of transition metal clusters is undoubtedly an important factor determining their physical and chemical properties[8]. Due to their increasing scientific and technological importance, considerable effort is currently devoted towards advancing our knowledge of their structure. An enormous effort has been invested to understand the size-dependent structural and physical properties of Ni clusters. The determination of the geometrical structure of transition metal clusters, i.e., how the atoms are packed together, remains an extremely difficult problem, both theoretically and experimentally. In order to understand these phenomena, one must know the structures of these small clusters. However, different authors have so far reported a wide range of structures. It is not clear, for example, at what size the cluster geometry changes from two-dimensional to three dimensional. Unfortunately, despite advances in experimental techniques, direct experimental determination of the atomic structure of small clusters is extremely difficult. Some indirect methods have to be used in detecting the structure of a cluster. For example, some of these properties have recently been studied by first principles(ab initio) calculations. KHANNA et al[4] provided information about the relationship between photoelectron spectroscopy and the magnetic moment of Ni7 clusters based on the density-functional theory and the generalized-gradient approximation. Although ab initio calculations provide significant insights into atomic bonding and some basic properties of lattice defects in Ni, many other properties and phenomena of interest can only be modeled correctly by using much larger simulation blocks and more sophisticated computer simulation methods such as molecular dynamics, Monte Carlo, genetic-algorithms.
Particle swarm optimization[9] was used to study structures and properties of atomic clusters as structural optimization technique, which has high efficiency and global search. In this work, a brief introduction to characteristic method as well as program design of particle swarm optimization was given at first. Then the structures and energies of Ni clusters were investigated using particle swarm optimization(PSO) combined with simulated annealing(SA) method[7, 10].
2 Computational methods
2.1 Particle swarm optimization
Particle swarm optimization is an evolutionary computation technique developed by EBERHART and KENNEDY in 1995[9], inspired partly by the social behavior of animal swarms (e.g. schooling fish and flocking birds), partly by human social behavior. The key idea is to have a swarm of interacting particles, each representing a candidate solution to a given optimization problem. Thus, particles are embedded in the search space and the solution space by ��flying�� around is explored. Moreover, instead of just randomly flying around, the particles are attracted to high-fit regions located by other particles. Each companion, called particle, in the population, which is called swarm, is assumed to ��fly�� over the search space in order to find promising regions of the landscape. In the function minimization case, such regions possess lower function values than others visited previously. In this context, each particle is treated as a point in a D-dimensional space that adjusts its own ��flying�� according to its flying experience as well as the flying experience of other particles. There are many variants of the PSO proposed so far, after EBERHART and KENNEDY[9] introduced this technique. In our experiments a version of this algorithm that is derived by adding an inertia weight to the original PSO dynamics was used.
First let us define the notation adopted in this work: the i-th particle of the swarm is represented by the D-dimensional vector Xi=(Xi1, Xi2, ��XiD), i=1,2, ��, N, and the best particle of the swarm, i.e., the particle with the smallest function value, is denoted by the index Pg=(Pg1, Pg2, ��, PgD), i=1, 2, ��, N. The best previous position of the i-th particle is recorded and represented as Pi=(Pi1, Pi2, ��, PiD), i=1, 2, ��, N, and the position change (velocity) of the i-th particle is Vi=Vi=(Vi1, Vi2, ��, ViD), i=1,2, ��, N. The particles are then manipulated according to the following equations:
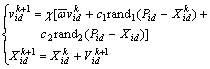 (1)
(1)
where d=1, 2, ��, D; i=1, 2, ��, N, and N is the size of swarm; c1 and c2 are two positive constants, here rand1 and rand2 are two random values into the range[0, 1]; �� is a constriction factor that is used in constrained optimization problems in order to control the magnitude of the velocity, here ��=0.729. The first equation is used to calculate the i-th particle��s new velocity by taking into consideration of three terms: the particle��s previous velocity, the distance between the particles best previous and its current position, finally, the distance between the swarm��s best experience and the i-th particle��s current position. Then, following the second equation, the i-th particle flies toward a new position. In general, the performance of each particle is measured according to a pre-defined fitness function, which is problem- dependent. The role of the inertia weight �� is considered very important in PSO convergence behavior. The inertia weight is employed to control the impact of the previous history of velocities on the current velocity, regulating the trade-off between the global and local exploration abilities of the swarm. A large inertia weight facilitates global exploration, while a small one tends to facilitate local exploration. A general rule of thumb suggests that it is better to initially set the inertia to a large value, in order to make better global exploration of the search space, and gradually decrease it to get more refined solutions, thus a linearly decreasing inertia weight value is used:
![]() (2)
(2)
where ��max=0.9, ��min=0.4. The method��s strength lies in its simplicity, being easy to code and requiring few algorithm parameters to define convergence behavior.
Simulated annealing method is used in particle swarm optimization. Among various theoretical algorithms, the simulated annealing method is believed to be the key to find the global minimum of a complicated system. Due to the inherent statistical nature of the simulated annealing, in principle, local minima can be hopped much more easily than that with many other methods.
2.2 Sutton-Chen potential
In the particle swarm optimization runs, we adopted the Sutton-Chen potential scheme for the description of the interaction between atoms[10]. The Sutton-Chen potential provides a reasonable description of various bulk properties, with an approximate many-body representation of the delocalized metallic bonding. However, it does not include any directional terms, which are likely to be important for transition metals with partially occupied d shells. The total energy of a system is expressed as
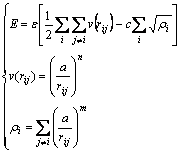 (3)
(3)
where c is a dimensionless parameter, �� is a parameter with dimensions of energy, a is the lattice constant, m and n are positive integers with n��m. The n, m and c parameters given by Sutton-Chen potential for Ni clusters are shown in Table1.
Table 1 Parameters of Sutton-Chen potential

3 Simulation results and discussion
The PSO-SA codes used were written by us. As a test of our computational method, we performed calculations to find the relation between the number of interactions and energy (the total cluster potential energy Vclu) for Ni5, as shown in Fig.1.
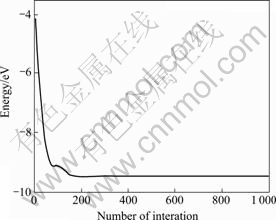
Fig.1 Energy as function of number of interactions for Ni5
From Fig.1, it��s found that the best member of the population improves rapidly in the first few generations, then there is a slower improvement, before the structure is found, at around 200 generations. This shows that the many-body potential provides a good and quick description of the bonding and energetics of the Ni cluster system. PSO differs from other evolutionary algorithms, which also shows that PSO-SA is good for the global optimization of clusters.
As a second test of our computational method, we performed calculations for Ni2, in which experimental data are available for comparison[11]. For the Ni2 dimer, the binding energy is obtained as 1.06 eV in our current PSO-SA results, which is in satisfactory agreement with the experimental data (Eb=1.04 eV).
With the above results in support of the reliability of our computational scheme, we next focused on the structures of Nin (n=2-19) clusters.
An unbiased global search for the lowest energy structures of Nin (n=2-19) clusters is performed in the PSO with Sutton-Chen potential. The PSO is an optimization strategy based on the behavior of animal swarms. Some stable structures of the Ni cluster obtained are shown in Fig.2.

Fig.2 Geometries of some stable structure of Nin (n=2-13, 19)
The structures obtained for Ni clusters in this size range are in good agreement with previous calculations [12-13], which shows that Ni clusters have structures based on icosahedral packing and the structures of all the clusters with the number of atoms n��6 are three- dimensional[14-17]. So the structural transition from two-dimensional(2D) structure to three-dimensional(3D) structure is found at around n=6. The calculated geometries of Nin are identical with those obtained by XIANG et al[6]. The present simulation gives the most stable structures for the clusters with the number 3-7 of atoms as an equilatriangle(3), tetrahedron(4b), trigonal bipyramid(5c), and pentagonal bipyramid(7a), respectively. For the clusters with the number 8-13 of atoms the pentagonal bipyramid keeps its form and additional atoms locate to form the icosahedral structure at n=13. For the 19-atom cluster, Ni19, a double-icosahedral structure forms. The structures of clusters seen from different angle for Nin (n=7, 11, 12, 13, 19) labeled as a and b in Fig.2. Ni5 has a trigonal bipyramid structure(5c) that is in agreement with experimental results[14]. The ground state of Ni7 agrees with that by SAROJ et al[3] and KHANNA et al[4]. It has been found that the five-fold symmetry is favored in the optimized structures. Five-fold symmetry structure starts with the seven-atom cluster, Ni7, which forms a pentagonal bipyramid. The structure of isomers of Ni8, bidisphenoid(point group D2d), agrees with that by SVEN et al[18], and the ground state of Ni13 agrees with that by GONG et al[10], which has the highest symmetry Ih.
The relative stability of the clusters described above can be studied by analyzing their energies. In order to obtain further general information on the size dependence of the structures, we first study the average interaction energy. The average interaction energy per atom E(E=|-En/n|) as a function of cluster size is shown in Fig.3. The simulation results indicate that the binding energy per atom tends to approach that of bulk materials when the atom number increases.
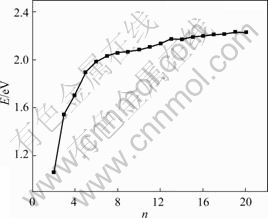
Fig.3 Average binding energy as function of Ni cluster size n
The structure stability of Nin clusters increases with the increase of total atom number n. It is also found that there exists direct correlation between the stability and geometrical structures of clusters, and relatively higher symmetry clusters are more stable. Then we study the difference in energy in adding an atom to the preceding cluster, that is, the first difference energy ��1En (��1En=En-En��1) and the second difference energy ��2En (��2En=En+1-2En+En��1). These energies are defined in terms of the total interaction energy of the cluster with the number of atoms n, respectively, and the ��2En is the difference of the energy of the two ��2En fragmentation paths: from Nin+1��Nin+Ni and Nin��Nin-1+Ni. If ��2En is positive, it means that the dissociation of Nin+1 into Nin leaving one atom free is more favorable than the dissociation of Nin into Nin+1, so ��2En is nothing but a measure of the stability of clusters. The second difference energy ��2En as a function of cluster size is shown in Fig.4. From it we can see that the clusters with the number of atoms 13 seem to be more stable. We may conclude that the common numbers in these groups of numbers might be the magic numbers for the Ni clusters studied in the present work. That is to say, the stability is not only size-dependent but also symmetrical structure- dependent.
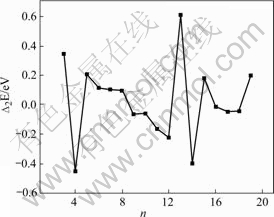
Fig.4 Second difference in binding energy as function of Ni cluster size n
It can be seen that there are some particular sizes having energetically magic characteristics. In order to obtain further general information on the size dependence of the structural growing, to confirm this magic behavior, and to further elucidate the growth trend of Ni clusters and the size evolution of Ni-Ni interactions, we describe the average bond length as a function of cluster size in Fig.5. We have calculated the average bond length using the following expression:
![]() (4)
(4)
where Rij is the distance between two atoms i and j. We consider an atom j to be the neighbor of atom i if their distance Rij is smaller than a cutoff of 0.252 nm, 4% longer than the bond length in bulk. Nb is the total number of bonds between atoms that lie below this cutoff.
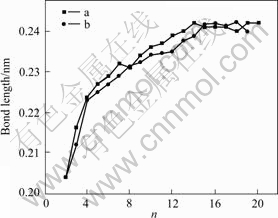
Fig.5 Bond length as function of Ni cluster size n (Curve a comes from Ref.[16] and curve b is our work)
The simulation results indicate that the bond length of Ni clusters increases with the increase of total atom number n and tends to approach that of bulk materials.
4 Conclusions
1) PSO differs from other evolutionary algorithms. The simulation results show that PSO-SA is good for the global optimization of clusters.
2) The structures of Ni clusters are icosahedral-like and the structural transition from two-dimensional(2D) structure to three-dimensional(3D) structure is found when n=6.
3) The binding energy per atom and the average bond length at various clusters sizes are obtained. The results show that they all tend to approach that of bulk materials when the number of atoms in the clusters increases. From the results of the second difference in the binding energy, the cluster at n=13 is more stable than others. The magic numbers effect is also found.
References
[1] WANG G H. Cluster physics [M]. Shanghai: Scientific and Technical Press, 2003. (in Chinese)
[2] MICHAELIAN K, Rendon N, Garzon I L. Structure and energetics of Ni, Ag, and Au nanoclusters [J]. Phys Rev B, 1999, 60(3): 2000-2010.
[3] Saroj K, Nayak B, Reddy B K, KHANNA S N, JENA P. Structure and properties of Ni7 cluster isomers [J].Chemical Physics Letters, 1996, 253: 390-396.
[4] Khanna S, Beltran M and Jena P. Relationship between photoelectron spectroscopy and the magnetic moment of Ni7 clusters [J]. Phys Rev B, 2001, 64(23): 235419(1-4).
[5] Luo C L. The structure of small nickel cluster: Ni2-Ni19 [J]. Modelling Simul Mater Sci Eng, 2000, 8: 95-101.
[6] Xiang Y, Sun D Y, Gong X G. Generalized simulated annealing studies on structures and properties of Nin (n=2-55) clusters [J]. J Phys Chem A, 2000, 104: 2746-2751.
[7] Xiang Y, Gong X G. Generalized simulated method and its application [J]. Progress in Physics, 2000, 20(3):319-334.
[8] Haluk M. Molecular dynamics simulations of super heated Ni14 [J]. Turk J Phy, 2000, 24: 607-615.
[9] Kennedy J, EBERHART R. particle swarm optimization [C]// Proc IEEE Int Conf on Neural Network. Perth, Australia: 1995. 1942-1948.
[10] Yao Y H, Gu X, JI M, Gong X G, WANG D S. Structures and magnetic moments of Nin (n=10-60) clusters [J]. Physics Letters A, 2007, 360: 629-631.
[11] Calleja M, Rey C, Alemany M M G, GALLEGO L J, ORDEJON P, SANCHEZ P D, ARTACHO E, SOLER J M. Self-consistent density-functional calculations of the geometries, electronic structures, and magnetic moments of Ni-Al clusters [J]. Phys Rev B, 1999, 60: 2020-2024.
[12] Papanicolaou N I, Chamati H, Evangelakis G A, PAPACONSTAN-TOPOULOS D A. Second-moment interatomic potential for Al, Ni and Ni-Al alloys, and molecular dynamics application [J]. Computational Materials Science, 2003, 27: 191-198.
[13] Teng Yu-yong, Zeng Xiang-hua, Zhang Hai-yan. Melting and glass transition for Ni clusters [J]. J Phys Chem B, 2007, 111: 2309-2312.
[14] Parks E K, Zhu L, Ho J, RILOY S J. The structure of small nickel clusters (I): Ni3-Nil5 [J]. J Chem Phys, 1994, 100: 7206-7222.
[15] ZHANG Q M, WELLS J C, GONG X G, ZHANG Zhen-yu. Adsorption of a carbon atom on the Ni38 magic cluster and three low-index nickel surfaces: A comparative first-principles study [J]. Phys Rev B, 2004, 69(20): 205413(1-7).
[16] Sun HQ, Ren Y, Wan G H. Equilibrium geometries of Nin (n=2-20) clusters [J]. Chinese Journal of Atomic and Molecular Physics, 2001, 18(4): 387-392.
[17] Wan X G, Zhou L, Dong J M, LEE T K, WANG D S. Orbital polarization, surface enhancement and quantum confinement in nanocluster magnetism [J]. Phys Rev B, 2004, 69: 174414.
[18] Sven K, Thomas J S, Alexander W, NOTKER R. Properties of isomers of the cluster Ni8 from density functional studies [J]. International Journal of Quantum Chemistry, 2000, 80: 567�C574.
Foundation item: Project (60371046) supported by the National Natural Science Foundation of China
Corresponding author: ZHOU Ji-cheng; Tel: +86-13873193957; E-mail: jicheng@mail.csu.edu.cn
