
���±�ţ�1004-0609(2011)08-1921-08
���Һ�м�����Ũ�ȶ����������Ʊ�TiO2����������ò������ܵ�Ӱ��
�� ��1�����ܾ�1, 2������ǫ1��������3
(1. ��������ҵ��ѧ �����о���Ժ�����Ͽ�ѧ�빤��ѧ�Ʋ������� 518055��
2. ���ϴ�ѧ ��ĩұ������ص�ʵ���ң���ɳ 410083��
3. ��������ҵ��ѧ �����о���Ժ ��������ľ����ѧ�Ʋ������� 518055)
ժ Ҫ��
�ֱ���0.5%HFˮ��Һ(A)��0.3 mol/L NH4F+70% H2O+30%������(�������)(B)��0.3 mol/L NH4F+30% H2O+70%������(�������)(C)��0.3 mol/L NH4F+3% H2O+97%��������Һ���Һ(�������)(D)��Ϊ���Һ���Ա��о������������Ե绯ѧ��������TiO2�������е���ò���ṹ������ܵ�Ӱ�졣��������� TiO2�������г�������������������Ӷ����ӣ�ˮ�����ҺA���Ʊ������ܳ��ȡ��ں�ܾ��ֱ�ԼΪ600��20��100 nm���ҷֲ������ȣ������������ҺD���Ʊ������ܳ��ȡ��ں�ܾ��ֱ�ԼΪ10 ��m��5 nm��60 nm���ҹܾ��ֲ����ȣ���Ʒ��O2�о�450 ���˻�2 h���ּ����������ҺA���Ʊ���TiO2����Ϊ�������ѿ�ṹ��������Ʒ��Ϊ�������ѿ���ͽ��ʯ��ṹ�����������ȵĽ������������TiO2�������еĺ�����Ӽ��ܱڼ�С���������Ч��������ߡ�
�ؼ��ʣ�
�绯ѧ�������������Һ��TiO2�����������
��ͼ����ţ�TQ153.6��TB34���� ���ױ�־�룺A
Effect of formamide in electrolyte on morphology and photocatalytic activity of TiO2 nanotube arrays by anodization
MA Qing1, LIU Shao-jun1, 2, WENG L��-qian1, DONG Wen-yi3
(1. Division of Materials Science and Engineering, Shenzhen Graduate School, Harbin Institute of Technology, Shenzhen 518055, China;
2. State Key Laboratory for Powder Metallurgy, Central South University, Changsha 410083, China;
3. Division of Urban and Civil Engineering, Shenzhen Graduate School, Harbin Institute of Technology, Shenzhen 518055, China)
Abstract: The influence of the fomamide concentration in the electrolytes on the morphology and structure was investigated in the electrolytes of 0.5%HF water solution (A), 0.3 mol/L NH4F+70%H2O+30% formamide (volume fraction) (B), 0.3% mol/L NH4F+30%H2O+70% formamide (volume fraction) (C) and 0.3 mol/L NH4F+3%H2O+97% formamide (volume fraction) (D). The nanotube becomes long and its wall thickness becomes smaller with increasing formamide concentration in electrolyte. The results show that the nanotube becomes longer and its wall thickness becomes smaller with increasing formamide concentration in electrolyte. The tube length, pore diameter and wall thickness of TiO2 nanotube arrays anodized in water-based electrolyte A are about 600 nm, 100 nm and 5 nm, respectively. The tube length, wall thickness and pore diameter for sample anodized in formamide-based electrolyte D are about 10 ��m, 5 nm and 60 nm, respectively. After being annealed at 450 �� for 2 h in oxygen, the single anatase structure is obtained for sample anodized in electrolyte A. In contrast, the mixed anatase and rutile phase are observed in TiO2 nanotube for the other three samples. The results of photodegradation of methyl orange (MO) show that TiO2 nanotubes with longer tube length and thinner wall thickness have higher photocatalytic activity.
Key words: electrochemical anodization; electrolyte; TiO2 annotube; photocatalytic
�������ṹ�Ķ�������(TiO2)����������������ͻ�ѧ�����Լ����õĻ�е�ͻ�ѧ�ȶ���[1]����̫���ܵ��[2]�������[3]������ԭ��[4]�Լ��������[5]�������й㷺��Ӧ��ǰ���������ܡ����ױ�Ĥ�����������Ϊ��ˮ�л��オ������ṹTiO2������У�����������Ż��ո��Ӽ������ظ���ʹ�õ����⡣�绯ѧ�����������Ʊ���TiO2�������в����������õĻ�еǿ��[6]�����Ҿ��и��ߵıȱ�������õĹ������[7]������õ��˸���Ĺ�ע��
���˵��ʱ��͵�ѹ���绯ѧ���������еĵ��Һ�����Ӱ��TiO2������ò��ṹ�Լ����ܵ���Ҫ����[8]��������������������������Ʊ�TiO2�������е�ˮ�Ե��Һ[9]�����Ʊ���TiO2���ܳ��Ƚ϶̣���ΪԼ500 nm�ҿ��ڶ�Բ�ζȽϲPAULOSE��[10]ʹ�ü����������Һ�����ڵķ�ˮ���л����Һ�Ʊ������ȳ���134 ��m�ij����������С����˵��ӽṹ�;���ṹ�������⣬����Ӱ���������ӵ������봫����TiO2���ܵij��ȡ��ܾ��Լ��ں����ò����Ҳ�Թ�����ܲ�����Ҫ��Ӱ�졣LAI��[1]������HF����Һ�����������Ʊ���TiO2���ܳ��ȼ���ᾧ�ȶ�������������ܵ�Ӱ�죬��û���ἰ���ܱں�Թ�����ܵ�Ӱ�졣LIU��[11]ʹ��0.5% HF�ͼ��������ĵ��Һ���������Ʊ��˳���Ϊ0.21 ��m��12 ��m��17 ��m��TiO2�������У�����17 ��m���ȵ�TiO2����������ʾ���õĹ�����ⱽ��Ч����Ȼ����LIANG��[12]����TiO2���ܵıں�����ܵij��ȶԹ�������и�Ϊ������Ӱ�죬����Լ28 nm�ں��������30 min���պ�ͻ��ԼΪ93%��2, 3-���ȱ��ӽ���Ч����
Ϊ��̽�����ܵij��Ⱥͱں����ò���ض���ת���Լ�������ܵ�Ӱ�죬�������߶Ա����۲�ͬ��ȵ�ˮ��������������Һ�Ե绯ѧ�����������Ʊ���TiO2�������еij��ȡ��ܾ��Լ��ں�ȱ�����ò��ṹ��Ӱ�졣���ڴ˻����ϣ��о�TiO2�������е���ò��ṹ�仯�Թ���������(Methyl orange, MO)�����Ե�Ӱ�졣
1 ʵ��
ʵ���ô��Ѳ�����Sigma-Aldrich(����99.7%�����0.25 mm)��ʵ��ǰ�ֱ��ñ�ͪ���Ҵ�������ˮ������ϴ���ں����и������0.5%HF���ˮ�Ի����Һ���绯ѧ�����������Ʊ�TiO2�������������� (25 ��)��20 V���ѹ�½��У���������ʱ��Ϊ1 h�����TiO2��������ǰ����������¯����2 ��/min����������������450 �沢����2 h��������¯��ȴ���˻�����Ϊ��������
TiO2���������Ĺ������ͨ���������������(MO)���ԣ�MO��ʼŨ��c0Ϊ20 mg/L�����ù�ԴΪPhilip 460 W��ѹ���ƣ�����������Ϊ365 nm����Դ����TiO2��������10 cm���������MO��Ӧ�����ƵĴ�ѭ����ȴˮϵͳ��ʯӢ�����н��С�����ǰ��ļ�������ֵ��ͨ���Ϻ�ݼ��723�Ϳɼ��ֹ��ȼƲⶨ����30��60��120��240 min��ȡ������������MO��463 nm����������ֵ��
����Hitachi S-4700������ɨ���������(FESEM)��JEM-2010(HR)���������(HRTEM)������������ò���б�����������ѧRigaku�߷ֱ�XRD��Renishaw Invia Micro-Raman Spectroscopy System���������Ƕ���������ṹ���б����������ձ�����Shimadzu UV-2450����-�ɼ��ֹ��ȼƶ�TiO2�������н�������-�ɼ����������չ���(UV-vis-diffuse reflection spectra, DRS)�ⶨ��
2 ���������
ͼ1��ʾΪ�ڲ�ͬ���Һ���Ʊ���δ���˻���TiO2�������е�SEM��1����Ϊ��ͬ���Һ���������Ʊ���TiO2����������ò�����ͽṹ�������ݡ���ͼ1(a)��(b)���Կ�����ˮ�����ҺA���Ʊ���TiO2���ܹܾ�ԼΪ50~100 nm���ܱں��ԼΪ15~20 nm�������ܿ��ڶ�Բ�ζȽϲ�����������ܱ�֮����ܽ����ʴ�������źܴ�̶ȵ�Сֱ�����ܺϲ�����Ϊ��ֱ�����ܵ��������ˣ����ܹܾ��ֲ������ȣ������ܵײ����н��ܡ����ܳ���ԼΪ600 nm�Ҳ��ֹܱڴ����ſ�����������(��ͼ1(b))����Ƚϣ����ŵ��Һ���л����������������ӣ����ܳ����������ӣ��ܾ���ںʼ�С���ơ������ҺD�м����������ﵽ��97%ʱ��TiO2���ܹܾ�ԼΪ60 nm�ҿ��ڶ˶�ΪԲ�ι�״���ܱں��ԼΪ5~10 nm�ҹܾ��ֲ�����(��ͼ1(g))��ͼ1(h)��ʾΪ�ڵ��ҺD�����������õ���TiO2�������еĵײ��Ͳ�����òͼ�����Կ���TiO2���ܵij������������ȳ�����10 ��m(��ͼ1(h))��ͬʱ�����Թ۲쵽TiO2����֮�����һ���ļ�϶�ҷֲ����ȣ�������ڸ�Ũ�ȼ������ĵ��Һ����������Զ��������ܡ���Ϊһ�ֱȽ��º͵ĵ��Һ[13]�������������Һ�еļ������ĵ����ʱ�ˮ�Ĵ�öࡣ��һ���棬���������Һ�������ṩ����ԴҲ��ˮ�����Һ��Ҫ�ٵö�[14]����Щ����һ���̶�����������������Ӧ��������F-��Ӧ����Ļ�ѧ�ܽ����ʣ��Ӷ�ʹ�ÿ�ʴ���ʽ����������ڸ���TiO2���ܵ��γɡ������ͨ�����������ܹ��ڸ�Ũ�ȼ������ĵ��Һ���Ʊ����ȵ�Ũ�ȼ��������Һ������TiO2���ܡ�ͼ2��ʾΪ��O2���վ�450 �����˻�2 h���TiO2�������е�SEM����ͼ2�ɼ����˻���TiO2�������нṹ�����ȶ��� û�з��������ı仯��

ͼ1 ��ͬ������Ũ�ȵ��Һ�Ʊ���TiO2�������е�SEM��
Fig.1 SEM images and cross-section images of as-anodized TiO2 nanotube arrays in electrolytes with different formamide contents
Ϊ�˽�һ��ȷ����������ò�ϵ�ϸ��𣬶��ڵ��ҺA��D���Ʊ������ֱܷ����HRTEM������ͼ3��ʾΪ�ڵ��ҺA��D���Ʊ���Ʒ���������պ�450 ���˻�2 h���HRTEM����ͼ3�ɼ����ڵ��ҺA���Ʊ�����Ʒ���˻����ƽ���ں�ԼΪ15~20 nm�����ܱں�Ȳ����ȣ��ܾ�ԼΪ90~100 nm���ڵ��ҺD���Ʊ�����Ʒ�������ܱ�Ҫ���Ծ����ҹܱ�ƽ����ƽ���ں�ԼΪ5~10 nm���ܾ�ԼΪ55~60 nm����Щ������1�е���ò��������һ�¡�

ͼ2 ��O2�����¾�450 ���˻�2 h��TiO2�������е�SEM��
Fig.2 SEM images of TiO2 nanotube arrays annealed at 450 �� for 2 h in oxygen
��1 ���Һ�м�����Ũ�ȶ�TiO2����������ò�ͽṹ��Ӱ��
Table 1 Effect of formamide content in electrolyte on morphologies and structures of TiO2 nanotube arrays

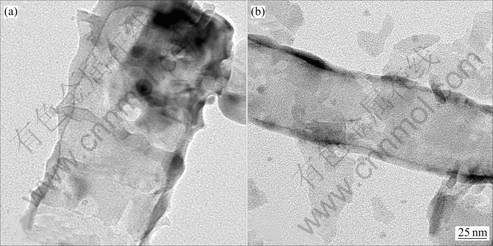
ͼ3 ������O2���վ�450 �����˻�2 h��TiO2���ܵ�HRTEM��
Fig.3 HRTEM images of TiO2 nanotube anodized in different electrolytes after annealed at 450 �� for 2 h in oxygen: (a) Electrolyte A; (b) Electrolyte D
ͼ4��ʾΪTiO2����������O2���վ�450 ���˻�2 h���XRD�ס�2�Ƚ�Ϊ25����48�㸽����������Ӧ�����ѿ������������壬��2�Ƚ�Ϊ27�㸽������������Ӧ�Ž��ʯ�����������壬����ΪTi�������������塣��ͼ4�ɼ�������TiO2���ܳ��ȵ����ӣ����ѿ����(101)�����ǿ��������ǿ���ڼ����������ҺD���Ʊ�����Ʒ�������ǿ��Ҫ���Ը�����A��B��C���Һ���Ʊ�����Ʒ�������ǿ�ȡ��������ˮ�����Һ�е������������ڼ��������Һ��������ʱ��TiO2���ܱڵĻ�ѧ��ʴ�����ᣬTiO2���ܵ�������ø����ȶ�����ˣ��Ʊ����� TiO2���ܳ���Զ����ǰ�߲�ʹ�����������ѿ���ĺ������ࡣ���ƵĽ����TiO2��ĤҲ��������[15]����ˮ�����ҺA��B��C���Ʊ���TiO2���ܾ�450 ���˻�����һ���ֽ��ʯ�࣬���ڼ����������ҺD���Ʊ���TiO2������450 ���˻����û�н��ʯ������ɡ����������еĽ��ʯ�����ѿ������������������ʽ(1)[16]��ã�
![]() (1)
(1)
ʽ�У�Ia��Ir�ֱ�Ϊ���ѿ�(101)�ͽ��ʯ(110)��������ǿ�ȣ�Kȡ0.79��
��ʽ(1)���Լ�������ҺA��B��C��D���Ʊ�����Ʒ�н��ʯ��ĺ����ֱ�Ϊ54.3%��1.7%��1.9%��0����Щ�����TiO2���ܱں���������еĹ�ϵ��VARGHESE��[17]������ƽ���ں�ԼΪ(27��6) nm�����������У������ܵ����ܱں�ߴ�����ƣ������ܱ���û�н��ʯ��ɺ˶�ֻ�����ܵײ������ĽӴ������н��ʯ�����ɡ�Ȼ����Ϊ�˳���ܱں������ѿ���ͽ��ʯ��֮�����ת�䶯��ѧ��ϵ���б�Ҫ���н�һ����ʵ�顣
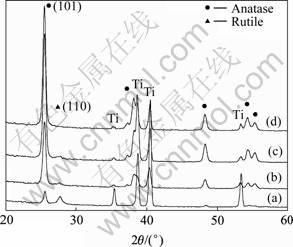
ͼ4 ��O2���վ�450 ���˻�2 h��TiO2��������XRD��
Fig.4 XRD patterns of TiO2 nanotube arrays anodized in electrolyte with different formide concentrations and annealed at 450 �� for 2 h in oxygen: (a) Electrolyte A; (b) Electrolyte B; (c) Electrolyte C; (d) Electrolyte D
�����XRD�ף�Raman�����Ƕ����ѿ���ͽ��ʯ���Ϊ���еIJ��ϱ����ֶΡ�ͼ5��ʾΪ��O2���վ�450 ���˻�2 h���TiO2��������Raman�ס��������ѿ��ͽṹTiO2�����Ӧ��Raman��ֱ�Ϊ147 cm-1 (Eg)��198 cm-1 (Eg)��398 cm-1 (B1g)��515 cm-1 (A1g+B1g)��640 cm-1 (Eg)[18]����ͼ5��ʾ���ڼ����������ҺD���Ʊ���TiO2����λ��146��199��395��517��638 cm-1����Raman�����Ӧ�����ѿ��࣬��û�н��ʯ����ij��֡���ˮ�����ҺA���Ʊ���TiO2����������444 cm-1������һ�����Ķ�Ӧ�Ž��ʯ���Eg(440 cm-1)�壬����������д����������Ľ��ʯ�࣬����XRD�Ľ����һ�¡����⣬������ڼ����������ҺD���Ʊ�����Ʒ�����ѿ���壬��ˮ�����ҺA���Ʊ�����Ʒ�����ѿ��������һ���̶ȵ����ơ����ַ�λ���ƶ�һ�������������ӳߴ�ЧӦ����ģ���һ����Ҳ�����ѿ�����ʯת��Ľ��[19]�����⣬�ڼ����������ҺD���Ʊ�����Ʒ��146 cm-1�������ѿ��������巢����ϸ�Ŀ���������������ھ��������ı�С�����������ͱ���ԭ����Ŀ�����ӣ�������ԭ������ȱ������ԭ�Ӷ���������ɳ�״̬��ʹ����Ӧ����Ƶ�ʱ���������[20]��
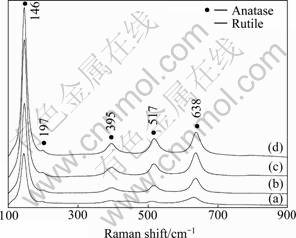
ͼ5 ��O2���վ�450 ���˻�2 h��TiO2������ Raman��
Fig.5 Raman spectra of TiO2 nanotube arrays anodized in electrolyte with different formide concentrations and annealed at 450 �� for 2 h in oxygen: (a) Electrolyte A; (b) Electrolyte B; (c) Electrolyte C; (d) Electrolyte D
ͼ6��ʾΪTiO2����������450 ����O2���˻�2 h���UV-vis-DRS�ס���ͼ6�ɼ�����ͬ������ܱڵ��������е�UV-vis�������߾������Բ�ͬ�IJ��ͨ��TiO2�������е����մ��߿���ֱ�����Ʒ��õ�[21]���ɴ˻�õ��ڵ��ҺA��B��C��D���Ʊ�����Ʒ��450 ���˻������մ��߷ֱ�Ϊ389��404��415��425 nm���������մ��ߵĺ����ܵ����������ò��Ӱ�죬����ɽ�ֱ��Ӱ�쵽TiO2�������е����մ���λ�á���TiO2������òҲ������մ��ߵ��ƶ�����һ����Ӱ�졣ZHUANG��[22]����2.5 ��m���ȵ�TiO2���ܽ�0.4 ��m���ȵ�TiO2���ܵ����մ��ߺ�����40~50 nm����ͼ6�����Կ����������������ҺD���Ʊ�����Ʒ�ڲ���С��500 nm�����ھ��и�ǿ�����գ�����Ҫ�����ڼ����������Һ���Ʊ�����Ʒ���и��������ܳ��ȣ�ʹ�����ڲ�������͵�λ����ܹ��������������Ŀ������[22]���������Uv-vis���ո�ǿ��LAI��[1]��������430 nm������С������Χ��UV-vis������Ҫ�����ڲ����Ѩ������ġ���Ƚϣ������ڼ����������ҺD���Ʊ�����Ʒ�ں����Խ��ͣ������̬�������Ӷ������˷Ƿ�������ı���������λ�ĸ��ϣ�������ܲ�����������Ŀ�����ӡ�
ͨ����TiO2����������450 ����O2���˻�2 h�������������MO����ѧ��������ɹ�ʽ(2)[23]�õ���������ͼ7��ʾ��
![]() (2)
(2)
ʽ�У�c0��c�ֱ�ΪMO�ij�ʼŨ�Ⱥʹ���Ӧ���Ũ�ȣ�A0��At��ֱ�Ϊ��ʼ��tʱ����պ�MO��463nm������ֵ����ͼ7��ʾ���հ���Ʒ(Blank)Ϊ��TiO2�����������������������������ߣ�k��2.15��10-4 min-1��ͨ����ϵ��ҺA��B��C��D���Ʊ���TiO2������������������MO���ߣ���kֵ�ֱ�Ϊ(4.03��0.209)��10-4 min-1��(6.28��0.208)�� 10-4 min-1��(8.68��0.708)��10-4 min-1��(5.06��0.528)�� 10-3 min-1�����Եأ����ҺD���Ʊ�����Ʒ��kֵԶ���ڵ��ҺA��B��C���Ʊ�����Ʒ��kֵ�������������������MOЧ��Ҫ������������3�������ġ������о�����[1, 11, 24]�������������ʵĸߵ���Ҫ��TiO2�������б�����ò����ṹ��Ӱ�졣һ���棬TiO2���ܵı�����������䳤�ȵ����Ӷ����ӣ���Ӧ�أ�Ҳ������TiO2�������MO�ĽӴ�����������ڹ�����ܵ���ߣ���һ���棬�����������ҺD���Ʊ���TiO2���ܱں��Ϊ5~10 nm��ԶС�ڵ��ҺA��B��C�����Ʊ���Ʒ�ıں�(Լ20 nm)�����ܱں�����ӻ��谭�����ӵĸ�ЧǨ��[24]��������������ԡ��ں�ļ�Сʹ��TiO2���ܱ���̬Ѹ�����ӣ������ڼ������Ӵ������ڲ�Ǩ�Ƶ����沢�����TiO2���ܵĹ������[12]��

ͼ6 ��O2�����о�450 ���˻�2 h��TiO2��������UV-vis-DRS����
Fig.6 UV-vis-DRS curves of TiO2 nanotube arrays anodized in electrolyte with different formide concentrations and annealed at 450 �� for 2 h in oxygen: (a) Electrolyte A; (b) Electrolyte B; (c) Electrolyte C; (d) Electrolyte D

ͼ7 TiO2������������������MO����ѧ�������
Fig.7 Kinetic curves of MO degradation of TiO2 nanotube arrays annealed in oxygen for 2 h: (a) Electrolyte A; (b) Electrolyte B; (c) Electrolyte C; (d) Electrolyte D
�����ڲ�ͬ���Һ���Ʊ���TiO2�������еij��Ȳ�ͬ�����о����������Խ����˳��ȵĹ�һ������������kֵ����TiO2���ܳ��ȡ���һ���� �������Ʒkֵ�ֱ�Ϊ(6.72��0.384)��10-4�� (6.78��0.384)��10-4��(2.48��0.202)��10-4��(5.06�� 0.0384)��10-4 min-1/��m�����ܵ��ҺA���Ʊ���TiO2�������г���ԼΪ0.60 ��m�������ҺD���Ʊ���TiO2�������г��������10 ��m���䳤��Ϊ���ҺA���Ʊ�������16���ࡣ���⣬���ҺD���Ʊ������ܱں�(5~10 nm)ҲԶ���ڵ��ҺA���Ʊ������ܱں�(Լ20 nm)������������������ȶ���ѧkֵ��Ϊ���ҺA���Ʊ�������Լ12������������Ч��û����TiO2���ܳ��ȳ��������ӵ�ԭ������������棺һ���棬�˻����ҺA���Ʊ�������Ϊ�������ѿ�ͽ��ʯ��ṹ��OHNNO��[25]��KAWAHARA��[26]���о�����������һ�������ĸ������ѿ�ͽ��ʯ��ṹ�У���������֮��ķ����ܼ�����ʹ��������洦�ܹ���Ϊ��������Ĵ��ڶ������ڹ������ӵIJ�����������Ч��Ҫ���ڵ������ѿ����ʯ��ġ���ˣ��������ѿ���ͽ��ʯ��Ĵ�����һ���̶����ֲ��˵��ҺA���Ʊ������������ܳ����϶�������Ч���IJ���Ӱ�졣��һ���棬��Ҳ����Ч��������Ӧ�����ܳ����йأ������ܳ��ȳ���һ����Χʱ���������ڹ�����ܵ���ߡ�������ܳ��ȳ�����������Ч���� ��[22]�������������յ���������ӽ�������Ϊ���ܳ��ȵ����Ӷ��������ӡ����⣬���ڼ��ȷ��Ӳ������������ܵ���ڣ���������ëϸ�����û����������ܵ��ڱڣ����ڱ��������ļ��ȷ�����Ŀͨ�������������ܳ��ȵ����Ӷ�����[12]��
3 ����
1) ���ŵ��Һ�м�����Ũ�ȵ����ӣ�ˮ��TiO2���ܵ��ܽ����ʴ�̶Ƚ��ͣ����TiO2���ܳ��ȵ������Լ����ڶ�Բ�ζȵ����ӡ�
2) ���ܱں��TiO2��ת������ҪӰ�죬��Ʒ��450 ���������˻�2 h�����������ҺD������������TiO2����(�ں�Ϊ5~10 nm)Ϊ�������ѿ�ṹ��������Ʒ(�ں�Լ20 nm)��Ϊ�������ѿ���ͽ��ʯ��ṹ��
3) TiO2�������е�������Ч��������TiO2�������еij��ȶ����������ӡ���TiO2�������еij��ȳ���һ����Χʱ�����������ڹ�����Ե���ߡ����ҺD���Ʊ�����Ʒ������������MOЧ��Ҫ������������3����Ʒ�ġ�Ȼ����ˮ�����ҺA���Ʊ�����������������Խϸߵĵ�λ����������������Ƚ����Խ��ʡ�
REFERENCES
[1] LAI Yue-kun, SUN Lan, CHEN Yi-cong, ZHUANG Hui-fang, LIN Chang-jian, CHIN J W. Effect of the structure of TiO2 nanotube array on Ti substrate on its photocatalytic activity[J]. J Electrochem Soc, 2006, 153(7): 123-127.
[2] O��REGAN B, GRATZEL M. A low-cost, high-efficiency solar cell based on dye-sensitized colloidal TiO2 films[J]. Nature (London), 1991, 353: 737-739.
[3] ASAHI R, MORIKAWA T, OHWAKI T, AOKI K, TAGA Y. Visible-light photocatalysis in nitrogen-doped titanium oxides[J]. Science, 2001, 293: 269-271.
[4] GRIMES C A. Synthesis and application of highly ordered arrays of TiO2 nanotubes[J]. J Mater Chem, 2007, 17: 1451-1457.
[5] YANG Bang-cheng, UCHIDA M, KIM H M, ZHANG Xing-dong, KOKUBO T. Preparation of bioactive titanium metal via anodic oxidation treatment[J]. Biomater, 2004, 25(6): 1003-1010.
[6] TSUCHIYA H, MACAK J M, TAVEIRA L, BALAUR E, GHICOV E, SIROTNA K, SCHMUKI P. Self-organized TiO2 nanotubes prepared in ammonium fluoride containing acetic acid electrolytes[J]. Electrochem Commun, 2005, 7(6): 576-580.
[7] ALBU S P, GHICOV A, MACAK J M, HAHN R, SCHMUKI P. Self-organized, free-standing TiO2 nanotube membrane for flow-through photocatalytic applications[J]. Nano Lett, 2007, 7(5): 1286-1289.
[8] RUAN C, PAULOSE M, VARGHESE O K, MOR G K, GRIMES C A. Fabrication of highly ordered TiO2 nanotube arrays using an organic electrolyte[J]. J Phys Chem B, 2005, 109: 15754-15759.
[9] MOR G K, VARGHESE O K, PAULOSE M, GRIMES C A. Transparent highly ordered TiO2 nanotube arrays via anodization of titanium thin films[J]. Adv Funct Mater, 2005, 15(8): 1291-1296.
[10] PAULOSE M, SHANKAR K, YORIYA S, PRAKASAM H E, VARGHESE O K, MOR G K, LATEMPA T A, FITZGERALD A, GRIMES C A. Anodic growth of highly ordered TiO2 nanotube arrays to 134 ��m in length[J]. J Phys Chem B, 2006, 110: 16179-16184.
[11] LIU Zhao-yue, ZHANG Xin-tong, NISHIMOTO S, JIN Ming, TRYK D A, MURAKAMI T, FUJISHIMA A. Highly ordered TiO2 nanotube arrays with controllable length for photoelectrocatalytic degradation of phenol[J]. J Phys Chem C, 2008, 112(1): 253-259.
[12] LIANG Hai-chao, LI Xiang-zhong. Effects of structure of anodic TiO2 nanotube arrays on photocatalytic activity for the degradation of 2,3-dichlorophenol in aqueous solution[J]. J Hazard Mater, 2009,162: 1415-1422.
[13] ��ع��, ������, ������. TiO2һά���ײ��ϼ������ṹ�ĺϳ�[J]. ��ѧ��չ, 2007, 19(4): 494-501.
ZHANG Yu-fang, ZHANG Zheng-guo, FANG Xiao-ming. Synthesis of one-dimensional TiO2 nanomaterials and their nanostructures[J]. Progress in Chemistry, 2007, 19(4): 494-501.
[14] CHRISTOPHERSEN M, CARSTENSEN J, VOIGT K, FOLL H. Organic and aqueous electrolytes used for etching macro- and mesoporous silicon[J]. Phys Status Solidi A, 2003, 197(1): 34-38.
[15] SHANG Jing, LI Wei, ZHU Yong-fa. Structure and photocatalytic characteristics of TiO2 film photocatalyst coated on stainless steel webnet[J]. J Molecula Catalysis A: Chemical, 2003, 202: 187-195.
[16] SPURR R A, MYERS H. Quantitative analysis of anatase-rutile mixtures with an X-ray diffractometer[J]. Anal Chem, 1957, 29: 760-762.
[17] VARGHESE O K, GONG Da-wei, PAULOSE M, GRIMES C A. Crystallization and high-temperature structural stability of titanium oxide nanotube arrays[J]. J Mater Res 2003, 18(1): 156-165.
[18] SWAMY V, KUZNETSOV A, DUBROVINSKY L S, CARUSO R A, SHCHUKIN D G, MUDDLE B C. Finite-size and pressure effects on the Raman spectrum of nanocrystalline anatase TiO2[J]. Physical Review B, 2005, 71: 184302.
[19] ������, ����, ������, �����. TiO2�����ӵ����ӳߴ�ЧӦ�����������[J]. ��ѧ��, 1999, 20(6): 613-618.
YU Xi-bin, WANG Gui-hua, LUO Yan-qing, LI He-xing. The quantum size effect and light absorption of ultrafine titania particles[J]. Chinese Journal of Catalysis, 1999, 20(6): 613-618.
[20] XU Jian-feng, CHENG Guang-xu, YANG Wei, DU You-wei. Raman scattering from surface phonons in fine Zn particles coated with ZnO[J]. J Phys B, 1996, 29(24): 6227-6232.
[21] RETUERT J, OUIJADA R, AREAS V. Porous titania obtained through polymer incorporated composites[J]. J Chem Mater, 1998, 10: 3929-3932.
[22] ZHUANG Hui-fang, LIN Chang-jian, LAI Yue-kun, SUN Lan, LI Jing. Some critical structure factors of titanium oxide nanotube array in its photocatalytic activity[J]. Environ Sci Technol, 2007, 41: 4735-4740.
[23] CHEN Ding-wang, RAY A K. Photodegradation kinetics of 4-nitrophenol in TiO2 suspension[J]. Wat Res, 1998, 32(11): 3223-3234.
[24] SREEKANTAN S, HAZAN R, LOCKMAN Z. Photoactivity of anatase-rutile TiO2 nanotubes formed by anodization method[J]. Thin Solid Films, 2009, 518: 16-21.
[25] OHNNO T, SARUKAWA K, TOKIEDA K, MATSUMURA M. Morphology of a TiO2 photocatalyst (Degussa, P-25) consisting of anatase and rutile crystalline phases[J]. J Catal, 2001, 203: 82-86.
[26] KAWAHARA T, KONISHI Y, TADA H, TOHGE N, NISHII J, ITO S. A patterned TiO2(anatase)/TiO2(rutile) bilayer-type photocatalyst: Effect of the anatase/rutile junction on the photocatalytic activity[J]. Angew Chem Int Ed, 2002, 41: 2811-2813.
(�༭ ����)
������Ŀ�����ҡ�ʮһ�塱�Ƽ�֧�żƻ�������Ŀ(2009ZX07423-04)��������ˮר�����(2008ZX07317-02)�������л����о��ƻ���Ŀ(JC200903120193A)�����ϴ�ѧ��ĩұ������ص�ʵ�鿪�Ż���������Ŀ
�ո����ڣ�2010-07-19�������ڣ�2010-10-28
ͨ�����ߣ����ܾ��������ڣ���ʿ���绰��0755-26032046��E-mail: shaojunliu@gmail.com
