J. Cent. South Univ. (2016) 23: 303-309
DOI: 10.1007/s11771-016-3074-4
Simultaneous separation and determination of four main isoflavonoids in Astragali Radix by an isocratic LC/ESI-MS method
WANG Yu-ling(������)1, 2, LIANG Yi-zeng(������)2, ZHANG Jie(�Ž�)2, FENG Xiao-liang(������)1,
GE Cheng-sheng(���ʤ)1, HUANG Lan-fang(������)2
1. Department of Chemical and Materials Engineering, Quzhou College, Quzhou 324000, China;
2. College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China
 Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Central South University Press and Springer-Verlag Berlin Heidelberg 2016
Abstract: A simple, reliable and rapid isocratic liquid chromatography (LC)-mass spectrometric detection (MS) coupled with electrospray ionization (ESI) method for simultaneous separation and determination of calycosin-7-O-��-D-glucoside, ononin, calycosin and formonometin in Astragali Radix was developed. After the samples were extracted with ethanol, the optimum separation conditions for these analytes were achieved using water and acetonitrile (70:30, v/v) containing 0.2% (v/v) acetic acid as a mobile phase and a 2.0 mm��150 mm Hypersil-Keystone C18 column. Selective ion monitoring (SIM) mode and [M+H]+ ions at m/z 447, 431, 285 and 269 were used for quantitative analysis of four main active components above mentioned. The calibration curves were linear in the range of 0.4-175.0 ��g/ml for calycosin-7-O-��-D-glucoside, 0.2-146.0 ��g/ml for ononin, 0.4-210.0 ��g/ml for calycosin and 0.5-217.0 ��g/ml for formonetion, respectively. The limits of quantification (LOQ) and detection (LOD) were 0.4 ��g/ml and 0.08 ��g/ml for calycosin-7-O-��-D-glucoside, 0.2 ��g/ml and 0.06 ��g/ml for ononin, 0.4 ��g/ml and 0.1 ��g/ml for calycosin, 0.5 ��g/ml and 0.1 ��g/ml formonetion, respectively. The standard recoveries were in the range of 96.5%-104.7%. The developed method has successfully been used for the determination of four main flavonoids in Astragali Radix from various sources and can be used for identification, differentiation and quality evaluation of Astragali Radix.
Key words: liquid chromatography; electrospray ionization (ESI); mass spectrometric detection (MS); isoflavonoids; Astragali Radix
1 Introduction
Traditional Chinese medicines (TCMs) have been extensively used to prevent and cure many diseases that have inflicted humans for many centuries due to the merits of low toxicity and rare complications. The pertinent researches include the analysis of active ingredients and major components of the medicine, treatment of diseases and the study for alternative drugs [1-2]. Astragali Radix, known as Huangqi in China and prepared from the dried roots of Astragalus membranaceus or A. membranaceus var. mongholicus, is a traditional Chinese medicine (TCM) and one of the most widely used Chinese herbs prescribed in many Chinese formulas to reinforce vital energy. Many Chinese doctors frequently use it as a tonic, an antiperspirant, a diuretic and for treatment of nephritis and diabetes [3]. Triterpene saponins, isoflavonoids, polysaccharides and biogenic amines are the known active constituents found in Astragali Radix [4-6]. Among them, isoflavonoids play an important role in human nutrition as health promoting natural chemicals and are established to be the beneficial components. It has been demonstrated that calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin are the major active isoflavonoids, although there are many other isoflavonoids in Radix astragali, such as isomucronulatol-7-O-��-D-glucoside, 9-methoxy-nissolin and isomucronulatol [7-10]. In particular, formononetin and calycosin, which are two most important active components in Astragali Radix, usually are considered indices for estimation of quality of Radix astragali and its products [10]. The structures of four main isoflavonoids mentioned above are shown in Fig. 1.
Over the past few years, natural products are a major resource in pharmaceutical industry. The research on guiding compounds for the possible new medicines, has been an increased interest. Because the separation process for active components from pharmaceutical plants with the traditional methods is very time-consuming, it is very important to develop highly effective, fast, sensitive and selective methods for primary screening and analysis of active components in natural plants. Only after we have confirmed the existence of useful components, further study becomes meaningful. There are several techniques for the analysis of isoflavonoids in Astragali Radix and its products, including spectrophotometry [11], high-performance liquid chromatography [12-17] and LC/MS-MS [18-20]. Reversed-phase HPLC is now commonly used method for the separation of complex mixtures of flavonoids in Astragali Radix. However, HPLC methods mentioned above suffer from limitation such as low sensitivity, poor selectivity due to the often-occurred low resolutions between flavonoids and low UV detection sensitivity. The reported LC/MS-MS methods present a high sensitivity. However, only limited components have been determined quantitatively. Also the experimented conditions reported so far do not give well separated individual peak. To the best of our knowledge, there are no reports on separation and determination of the four active isoflavonoids in Astragali Radix by an isocratic LC/MS. The purpose of the present work is to develop a simple, rapid, selective and sensitive isocratic LC/MS method for simultaneous separation and determination of active components in Astragali Radix. Based on this work, the contents of four main bioactive isoflavonoids in Astragali Radix from various sources have been determined and compared.
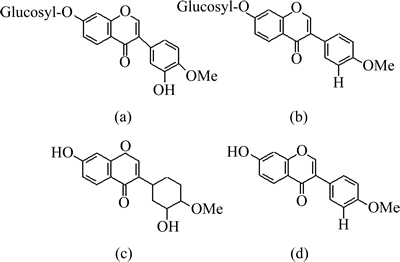
Fig. 1 Structures of calycosin-7-O-��-D-glucoside (a), ononin (b), calycosin (c) and formonometin (d)
2 Experimental
2.1 Instrumentation
All experiments were carried out on a Shimadzu LCMS-2010 equipment (Kyoto, Japan), which includes a LC-10Advp solvent delivery pump, a FCV-10ALvp low pressure gradient unit, a DGU-14A degasser, a CTO-10Avp column oven, a SPD-M10Avp photodiode array detector, a quadrupole mass spectrometer. R2003KE rotary evaporator was manufactured by Shanghai Senco Technology Co., Ltd. (Shanghai, China).
2.2 Materials and standards
The plant materials used in this work were purchased from main wholesale traditional Chinese medicines markets and main city pharmacy in China and were authenticated by a researcher from Institute of Herbal Drugs, Hunan academy of Traditional Chinese Medicine. Reference compounds of calycosin-7-O-��-D- glucoside, ononin, calycosin and formonometin were purified from A. membranaceus root extracts and characterized by 1H-NMR and 13C-NMR spectroscopy. The purity of the four reference compounds was also evaluated by LC/ESI-MS. Purity was found to be greater than 96% in all cases. 1.0 mg/ml stock solution of each component including calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin was prepared in methanol. Working standard solution (10 ��g/ml) of each component was prepared by diluting the stock solution with methanol. All solutions were filtered through a 0.45 ��m membrane and degassed and then were stored in a refrigerator (at 4 ��C) and brought to room temperature before use. HPLC-grade acetonitrile was obtained from Hanbang Science and Technology Co. (Jiangsu, China). Water was double distilled and subsequently filtered through a 0.45 ��m membrane (Millipore, Bedford, MA, USA). Other regents were of analytical grade.
2.3 Sample preparation
The roots of Astragali Radix were powdered to a homogeneous size and sieved through a No.40 mesh. The obtained powder samples were placed in an oven and dried at 40 ��C for 4 h. 2.0 g of ground power was refluxed with 200 ml methanol-water (80:20, v/v) for 3 h, then the methanol raw extract was filtered and the filtrate was evaporated to dryness in vacuum. The viscous residue was rotated in 25 ml hot water and the suspension was extracted twice with 10 ml and 5 ml ethyl acetate. The ethyl acetate phases were combined, and ethyl acetate was evaporated to dryness in vacuum. The residue was dissolved in 2.0 ml methanol. A 5 ��l of the sample solution filtered with 0.22 ��m membrane was injected into the HPLC column.
2.4 Chromatographic conditions
The column utilized for separation was a 2.0 mm��150 mm Hypersil-Keystone C18 column with a particle size of 5 ��m. The analytical column was protected by a C18 guard-pak cartridge (Waters, Milford, MA, USA). The mobile phase consisted of water and acetonitrile (70:30, v/v) containing 0.2% (v/v) acetic acid. Each component of the mobile phase was filtered through a 0.22 ��m membrane. All separations were at room temperature and a flow-rate of 0.2 ml/min. The amount of injection was 5 ��l. The HPLC-DAD chromatographic profile was recorded at 260 nm.
2.5 Mass spectrometric detection conditions
The ESI-MS spectra were acquired in the positive ion and scan mode using an electrospray interface. The positive selective ion monitoring (SIM) mode and [M+H]+ at m/z 447, 431, 285, 269, which was selected as the SIM ion, were chosen for quantification of the four main isoflavonoids, calycosin-7-O-��-D-glucoside, ononin, calycosin and formonometin. The ionization parameters for both scan and SIM mode were as follows: ESI temperature was 345 ��C; Curved desolvation line (CDL) and block temperature were 230 ��C and 210 ��C, respectively; Probe voltage was 4.5 kV; Detector voltage was 1.8 kV; CDL voltage was -16 V; Q-array Bios was 42 V; Nebulizing gas flow was 4.5 L/min.
2.6 Validation of quantitative method
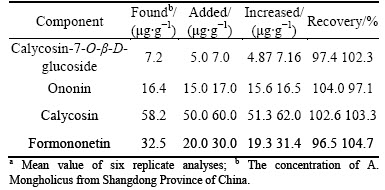
By serial dilution of the working and stock solution of each component with methanol, calibration standards at levels of 0.4, 2.0, 5.0, 20.0, 50.0, 100.0 and 175.0 ��g/ml for calycosin-7-O-��-D-glucoside, 0.2, 1.0, 5.0, 20.0, 50.0, 100.0 and 146.0 ��g/ml for ononin, 0.4, 2.0, 5.0, 20.0, 50.0, 120.0 and 210.0 ��g/ml for calycosin, 0.5, 2.0, 5.0, 20.0, 50.0, 120.0 and 217.0 ��g/ml for formonetion, were obtained. Each calibration standard was determined three times. The standard curves were calculated by plotting peak area ratio (Y) of each analyte in total ion chromatogram (TIC) of LC/ESI-MS versus concentration (X, ��g/ml) with least squares linear regression. The retention times of calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin in LC/ESI-MS TIC were 6.88 10.75 12.74 18.57 min, respectively. The method has been validated for selectivity, linearity, precision, accuracy and recovery. Quality control samples of each analyte at concentrations of 1.0, 40.0 and 140.0 ��g/ml were prepared by diluting the stock and working solutions with the mobile phase. The precision and accuracy were determined by six replicate analyses of quality control samples. 12.0 ��g calycosin-7-O-��-D- glucoside, 40.0 ��g ononin, 120.0 ��g calycosin and 80.0 ��g formononetin at sample level were added to 2.0 g A. mongholicus powder from Shangdong Province of China and performed as sample preparation. This spiked sample was also used to evaluate precision and accuracy. Various references of calycosin-7-O-��-D- glucoside, ononin, calycosin and formononetin at sample level (see Table 3) were added to 2.0 g actual samples. after processing as sample preparation, the concentration of these components was determined and recoveries were calculated. The recovery was evaluated by comparing the peak area response of analytes in extracted samples and standard added samples.
3 Results and discussion
3.1 Optimization of separation conditions
The separation of iso-flavonoids was very difficult due to their similar structure (see Fig. 1). Then, optimizing the chromatographic conditions was essential. Some reports used the isocratic HPLC methods, but the components which were to be determined overlapped heavily with other components exist in the actual sample (see Fig. 2(b)) [9]. In these reports, the quantitative analysis of the components was based on peak area or height, but it would be difficult to determine the areas of the overlapped peaks. So, the results obtained by what has been mentioned above were questionable. WU et al [14] reported a gradient elution system for the separation and determination of some main isoflavonoids in Radix Astragali. However, the separation was as long as 40 min, not including the equilibration time between runs. In a preliminary study, it was found that separation of four main iso-flavonoids was achieved on a 2.0 mm��150 mm Hypersil-Keystone C18 column by using the mobile phase consisting of water, acetonitrile and acetic acid. Separation conditions such as percentage of water, acetonitrile, acetic acid were optimized with the reference solution. The optimum chromatographic conditions were summarized in Section 2.4. The full peak-to-baseline resolution of the four major isoflavonoids was achieved under the chromatographic conditions developed in this work. The HPLC-DAD profile of the reference solution was monitored at 260 nm (see Fig. 2(a)). The mass concentration of each reference was 30 ��g/ml. 1, 2, 3 and 4 were calycosin- 7-O-��-D-glucoside, ononin, calycosin and formonometin, respectively. The retention time (tR) and UV data of each component are summarized in Table 1.
3.2 Optimization of ESI-MS conditions
Although the mixture of four reference compounds can be separated at the optimal chromatographic conditions, it was quite difficult to determine calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin simultaneously with HPLC-DAD because of the presence of other components in Astragali Radix with different polarities and consequently, different retention times. Furthermore, some components with very low contents in real samples can not be detected with HPLC-DAD due to the low inherent UV absorbance and concentration of these components (see Fig. 2(b)).
Here a rapid isocratic LC/MS method based on coupling with the ESI interface was developed. On-line molecular mass information in the analysis of flavonoid references was provided by the use of LC/ESI-MS. In order to obtain optimum ionizing conditions, using the reference solution, both an atmospheric pressure chemical ionization (APCI) and ESI interface were tested in positive and negative ion mode by scanning from m/z 200 to 500. The base peak intensities of positive ion were higher than those of negative ion in both ionization modes. ESI mode exhibited a more intensive peak than APCI mode, so the positive ion ESI mode was selected in this study. The optimum ESI-MS conditions were listed in Section 2.5. At the optimal chromatographic conditions and ESI-MS conditions, mass spectrum and the MS data of each reference are obtained. The MS data of each reference were also listed in Table 1.
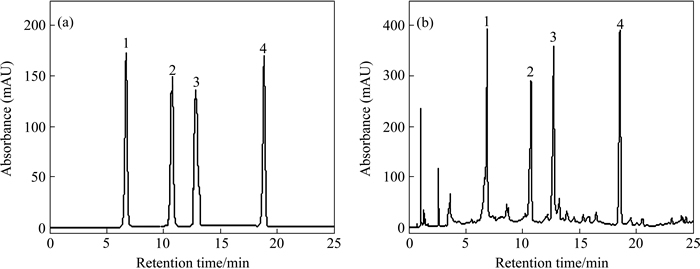
Fig. 2 HPLC chromatograms of reference solution (a) and methanol extract of A. mongholicus from Ganshu Province, China (b)
Table 1 values of tR, UV ��max, [M+H]+ and [M+Na]+ of flavonoid references
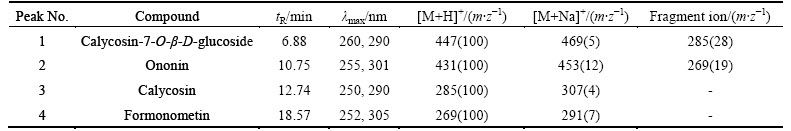
As listed in Table 1, each of the references showed significant and distinctive [M+H]+or [M+Na]+ion, UV spectra, and quite different retention time. Consequently, these characterizations of standards can be used for comparison with the chromatograms of various resources of Astragali Radix extracts. Each reference was characterized by a protonated molecular ion [M+H]+ (at m/z 447, 431, 285 and 269, respectively) as base peak. So, SIM mode involving the use of the [M+H]+ ions at m/z 447, 431, 285 and 269 was chosen for simultaneous determination of the four active components.
A mixture containing four references was injected into the LC/MS system. They were monitored using SIM mode. TIC of calycosin-7-O-��-D-glucoside, ononin, calycosin and formonometin reference mixture in SIM mode is shown in Fig. 3(a). The mass concentration of each reference was 30 ��g/ml and peaks were identified as shown in Fig. 2. There were fragment ions in MS data of calycosin-7-O-��-D-glucoside and ononin, which were consistent with their structure due to removing its related glycoside. This was also helpful for the identification of flavonoid in Astragali Radix extract. UV spectrum and mass spectrum of each reference were used for comparison with the chromatograms of Astragali Radix extracts from different locations for identification of components.
3.3 Validation of calibration curves, linearity and detection limit
Calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin, the most important biologically active components in Astragali Radix, are very important for primary screening and analysis and quality control of this commonly used drug. Selective ion monitoring (SIM) mode ([M+H]+ at m/z 447, 431, 285 and 269 was used for quantitative analysis of above four active components. The calibration curves were linear in the studied range of 0.4-175.0 ��g/ml for calycosin-7-O-��-D-glucoside, 0.2-146.0 ��g/ml for ononin, 0.4-210.0 ��g/ml for calycosin and 0.5-217.0 ��g/ml for formonetion, respectively. The calibration curves were Y=1.486��104X-3.251��103, Y=1.674��104X+3.442��103, Y=1.428��104X+ 3.173��103 and Y=1.561��104X+2.264��103 with correlation coefficients of 0.9992, 0.9984, 0.9975, and 0.9985 for calycosin-7-O-��-D-glucoside, ononin, calycosin and formonetion, respectively. The good selectivity was essential to an analytical method and the most important aim was checked so that there was no interference between the four isoflavonoids and the components of the biological matrix. No interference in the retention time areas of four isoflavonoids was observed due to using SIM mode in this method. Under these conditions, four isoflavonoids showed favorable selectivity. Because Astragali Radix contained analyte, no real blank was available for preparation of standards or controls. A solvent blank was analyzed for determining limit of detection (LOD). The limits of detection were 0.08 ��g/ml for calycosin-7-O-��-D- glucoside, 0.06 ��g/ml for ononin, 0.1 ��g/ml for calycosin and formonetion, which were determined from signal-to-noise ratio of 3:1. The lower limit of the range of the calibration curve can be considered the limits of quantification of this method. The limits of quantification of this method were 0.4 ��g/ml for calycosin-7-O-��-D-glucoside, 0.2 ��g/ml for ononin, 0.4 ��g/ml for calycosin and 0.5 ��g/ml for formonetion.
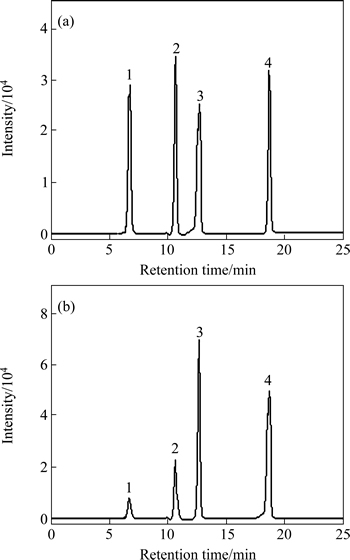
Fig. 3 LC/ESI-MS total ion chromatograms (TIC) of reference solution (a) and methanol extract of A. mongholicus from Shangdong Province, China (b)
3.4 Validation of precision, accuracy and recovery
The relative standard deviations of retention times of calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin were 0.27, 0.24, 0.18 and 0.13, respectively. Six replicate analyses of quality control samples and a spiked actual sample were used to calculate the precision and accuracy. The results are listed in Table 2. The accuracy was in the order of -6.0%-3.8% for calycosin-7-O-��-D-glucoside, -2.0%-5.0% for ononin, -4.0%-3.0% for calycosin and -2.8%-4.0% for formononetin. The precision was in the order of 1.8%-5.7% for calycosin-7-O-��-D-glucoside, 1.6%-6.4% for ononin, 1.4%-5.8% for calycosin and 1.5%-6.1% for formononetin. Various concentrations of standard solutions of calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin were added to 2.0 g A. mongholicus from Shangdong Province of China and recoveries were determined using the conditions described above. The results are shown in Table 3. The recoveries were 96.5%-104.7%. As listed in Tables 2 and 3, recovery, precision and accuracy of the developed method were very satisfactory. Obviously, the developed method has a very wide linear range, good selectivity, low limits of detection and quantification.
Table 2 Summary of precision and accuracy
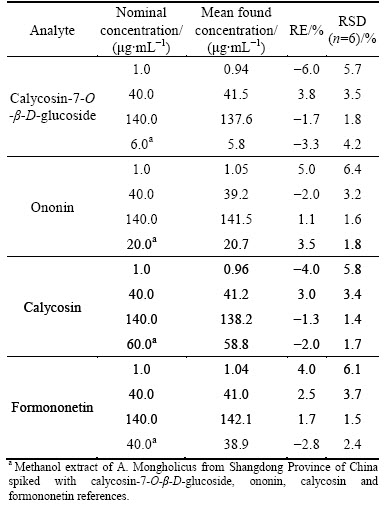
Table 3 Recoveries of calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetina
3.5 Stability
Each isoflavonoid reference at concentrations of 1.0, 40.0 and 140.0 ��g/ml (low, middle and high) were used for stability experiments. The results indicated that the difference of measured concentration from time 0 to 10 h was less than 5.1% when these samples were placed at room temperature, which allowed us to conclude that processed samples were stable for at least 10 h. In long-term stability experiments, after storage for 20 d at 4 ��C, more than 96.4% of each isoflavonoid reference remained according to their peak areas at each concentration. This demonstrated that these isoflavonoid references were stable for at least 20 d when they were stored at 4 ��C.
Stability of a processed sample was also performed. Processed A. mongholicus from Shangdong Province of China was used for study of stability. As shown in Table 4, the concentrations of calycosin-7-O-��-D- glucoside, ononin, calycosin and formononetin were 7.2, 16.4, 58.2 and 32.5 ��g/g, respectively. The sample was analyzed every 4 h at ambient temperature. The results are listed in Table 4. The results showed that these isoflavonoids in the processed sample were stable for at least 12 h at room temperature if the allowed difference between the found and nominal concentration were 5%. After they were stored for one month at 4 ��C, more than 95% of these four isoflavonoids remained. This showed that these four isoflavonoids in processed samples were stable for at least 30 d when it was stored at 4 ��C.
3.6 Application
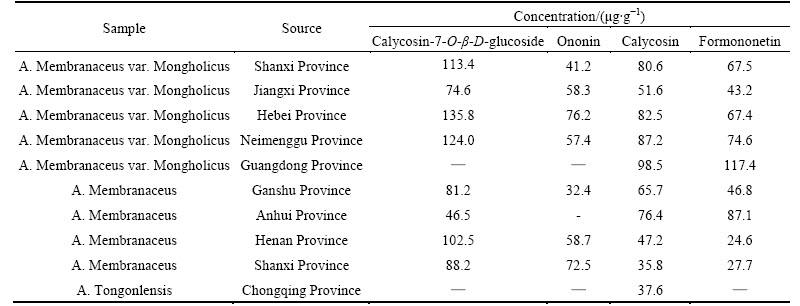
The developed method has been applied to quantitative determination of calycosin-7-O-��-D- glucoside, ononin, calycosin and formononetin in Astragali Radix. A typical LC/ESI-MS TIC of A. mongholicus from Shangdong Province of China was shown in Fig. 3(b). The determined results are listed in Table 3. The concentrations of calycosin-7-O-��-D- glucoside, ononin, calycosin and formononetin in Astragali Radix from many other sources were also determined and compared. The results are listed in Table 5. As listed in Table 5, as for the concentration of each isoflavonoid, no obvious differences existed in two main Astragali Radixs, A. membranaceus var. mongholicus and A. membranaceus. The concentrations of these four main isoflavonoids in A. membranaceus var. mongholicus were somewhat higher than differences from locality to locality. The method can be used to identify the species of Astragali Radix and differentiate its false or substitute, which has not been medicinally used by China Pharmacopoeia. However, the concentration of each isoflavonoid in Astragali Radix showed obvious a sample from Chongqing was identified as A. tongonlensis, which was not A. membranaceus var. mongholicus or A. membranaceus. Since isoflavonoids are important biologically active components, quality control of this commonly used drug is in great demand. Determination of the four main isoflavonoids can be of great importance for the identification, differentiation and quality evaluation of Astragali Radix.
Table 4 Stability of calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin in a processed sample
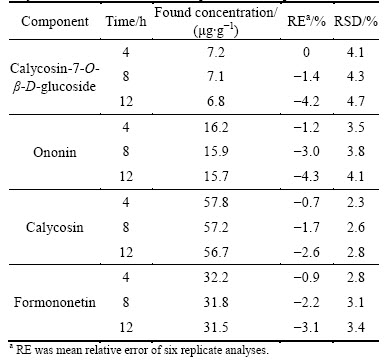
Table 5 determination results of calycosin-7-O-��-D-glucoside, ononin, calycosin and formononetin in Astragali Radix from different source
4 Conclusions
1) A sensitive, fast, accurate and selective isocratic liquid chromatographic-electrospray ionization mass spectrometric method for the determination of four main isoflavonoids in Astragali Radix is developed. The mass spectra of isoflavonoids obtained from the extracts of actual samples are characterized by [M+H]+ as base peak. SIM mode is used for quantitative analysis of four active components.
2) The developed method can be used for differentiation and quality evaluation of the widely used Astragali Radix from different sources.
References
[1] ZHONG X, YAN J, LI Y C, KONG B, LU H B, LIANG Y Z. A novel strategy for quantitative analysis of the formulated complex system using chromatographic fingerprints combined with some chemometric techniques [J]. Journal of chromatography A, 2014 1370(1): 179-186.
[2] XU Xiao-na, JIANG Jun-hui, LIANG Yi-zeng, LI Xiao-ru, YI Lun-zhao, CHENG Jin-le. Chromatographic fingerprint analysis of Fructus Aurantii Immaturus by HPLC-DAD and chemometric methods [J]. Journal of Central South University of Technology, 2011, 18(2): 353-360.
[3] LIU De-li, BAO Hua-yin, LIU Yang. Progress on chemical constituents and pharmacological effects of Astragali Radix in recent five years [J]. Food and Drug, 2014, 16(1): 68-70. (in Chinese)
[4] KWON H J, PARK Y D. Determination of astragalin and astragaloside content in Radix Astragali using high-performance liquid chromatography coupled with pulsed amperometric detection [J]. Journal of Chromatography A,2012, 1232(2): 212-217.
[5] SUN Jie, ZHANG Lei, ZHANG Xiao-long, YAN Rui-qing, CHAI Xin, WANG Yue-fei. Chemical constituents from A. membranaceus var. mongholicus [J]. Drugs & Clinic, 2013, 28(2): 138-143. (in Chinese)
[6] WU Hong-wei, FANG Jing, TANG Li-ying, LU Peng, XU Hai-yu, ZHAO Ye, LI De-feng, ZHANG Yi, FU Mei-hong, YANG Hong-jun. Quality evaluation of Astragali Radix based on DPPH radical scavenging activity and chemical analysis [J]. Chinese Herbal Medicines,2014, 6(4): 282-289. (in Chinese)
[7] ZHAO B S, FU Y J, WANG W, ZU Y G, GU C B, LUO M, EFFERTH T. Enhanced extraction of isoflavonoids from Radix Astragali by incubation pretreatment combined with negative pressure cavitation and its antioxidant activity [J]. Innovative Food Science & Emerging Technologies,2011, 12(4): 577-585.
[8] DONG X R, LIANG Y Z, WANG B, LONG X H. Simultaneous separation of four Flavonoids and two Astragalosides from radix Astragali by semi-preparative LC [J]. Chromatographia, 2010, 71(3/4): 225-231.
[9] XIAO W H, HAN L J, HI B. Microwave-assisted extraction of flavonoids from Radix Astragali [J]. Separation and Purification Technology, 2008, 62: 614-618.
[10] ZHOU Chao, HE Yi, LU Jing, LIN Rui-chao, BI Kai-shun. Studies on the preparation of isoflavones reference extract and application on the assay of Astragalus Radix [J]. Chinese Journal of Pharmaceutical Analysis, 2014, 34(3): 523-527. (in Chinese)
[11] SONG Xin-yue, LI Ying-dong, SHI Yan-ping, JIN Ling, CHEN Juan. Quality control of traditional Chinese medicines: a review [J]. Chinese Journal of Natural Medicines,2013, 11(6): 596-607. (in Chinese)
[12] SONG J Z, YIU H W,QIAO C F,HAN Q B,XU H X. Chemical comparison and classification of Radix Astragali by determination of isoflavonoids and astragalosides [J]. Journal of Pharmaceutical and Biomedical Analysis, 2008, 47(2): 399-406.
[13] CHENG Hai-yan, CHEN Xiao-hui, LI Qing, TAN Xiao-jie, WANG Peng, BI Kai-shun1. RP-HPLC simultaneous determination of six flavonoids in Radix Astragali [J]. Chinese Journal of Pharmaceutical Analysis, 2009, 29(7): 1115-1118. (in Chinese)
[14] WU T, ANNIE-BLIGH S W, GU L H, WANG Z T, LIU H P, CHENG X M, BRANFORD-WHIT C J, HU Z B. Simultaneous determination of six isoflavonoids in commercial Radix Astragali by HPLC-UV [J]. Fitoterapia, 2005, 76(2): 157-165.
[15] ZHOU Qian, LI Ying,DAI Yan-peng,SHEN Xiu-juan, SHUN Li-li. Simultaneous determination of four isoflavonoids from Astragali Radix and Yupingfeng Granules [J]. Chinese Traditional Patent Medicine, 2014, 36(8): 1674-1677. (in Chinese)
[16] SONG Xiao-wei, LI Qing, YE Jing, ZHANG Yuan-ting, CHEN Xiao-hui, BI Kai-shun. Comparison of flavonoid components in Astragali Radix and its processed products [J]. Chinese Journal of Experimental Traditional Medical Formulae, 2013, 19(9): 85-88. (in Chinese)
[17] SUN Lian-xia, CHEN Zhi-hong, GAO Wei-zhuan. Study progress on the determination of flavonoids in Astragali Radix by high-performance liquid chromatography [J]. Journal of Chengde Medical College, 2014, 31(1): 62-65. (in Chinese)
[18] QI L W, CAO J, LI P, YU Q T, WEN X D, WANG Y X, LI C Y, BAO K D, GE X X, CHENG X L. Qualitative and quantitative analysis of Radix Astragali Products using rapid resolution liquid chromatography-diode array detection coupled with time-of-flight mass spectrometry with dynamic adjustment of fragmentor voltage [J]. Journal of Chromatography A, 2008, 1203(1): 27-35.
[19] QI L W, LI P,REN M T, YU Q T, WEN X D, WANG Y X. Application of high-performance liquid chromatography-electrospray ionization time-of-flight mass spectrometry for analysis and quality control of Radix Astragali and its preparations [J]. Journal of Chromatography A, 2009, 1216(11): 2087-2097.
[20] LI M H, TONG X, WANG J X, ZOU W, CAO H, SU W W. Rapid separation and identification of multiple constituents in traditional Chinese medicine formula Shenqi Fuzheng Injection by ultra-fast liquid chromatography combined with quadrupole-time-of-flight mass spectrometry [J]. Journal of Pharmaceutical and Biomedical Analysis,2013, 74(1): 141-155.
(Edited by YANG Hua)
Foundation item: Project(21472110) supported by the National Natural Science Foundation of China; Project(LY15B050008) supported by the Natural Science Foundation of Zhejiang Province, China; Project(2013Y003) supported by Quzhou Technology Projects, China
Received date: 2014-12-26; Accepted date: 2015-05-05
Corresponding author: GE Cheng-sheng, Professor; Tel: +86-570-8015339; E-mail: gechengshen@qzu.zj.cn

