DOI: 10.11817/j.ysxb.1004.0609.2020-35831
单晶/多晶镍复合体剪切过程分子动力学模拟
李源才,江五贵,周 宇
(南昌航空大学 航空制造工程学院,南昌 330063)
摘 要:采用整体叶盘技术将不同性能指标的单晶镍涡轮叶片和多晶镍盘片连接为一个整体,然而却带来盘叶连接区断裂失效问题,严重制约了该技术的发展。为此,本文采用分子动力学研究单晶/多晶镍复合体的剪切性能。首先讨论了不同晶态镍的剪切性能,然后重点研究了单晶/多晶镍复合体的剪切速率效应与温度效应。结果表明:相比于单晶镍,由于多晶镍的加入,单晶/多晶镍复合体的整体剪切应力水平有所下降,其剪切强度显著小于单晶镍的剪切强度。不同晶态镍剪切变形进入塑性阶段后,出现类似于宏观材料颈缩的现象,靠近颈缩区的原子发生局部错动、重排以及部分非晶化。随着剪切速率的增加,单晶/多晶镍复合体的剪切模量逐渐增大。随着温度升高,单晶/多晶镍复合体剪切模量却呈现下降趋势。
关键词:整体叶盘;单晶/多晶镍复合体;分子动力学;剪切速率效应;温度效应
文章编号:1004-0609(2020)-08-1837-09 中图分类号:TB31 文献标志码:A
采用航空发动机整体叶盘先进连接技术可以将不同性能指标的叶片(耐蠕变)和盘片(抗疲劳)连为整体,然而却使得盘叶结合区成为整个部件的薄弱区域[1-2]。在部件制备过程中引入的缺陷,对于强调高可靠性和长寿命的航空发动机高速转动部件来说是个重要隐患。因此,结合区的力学性能成为决定双晶态整体叶盘能否得到广泛应用的关键因素。采用计算机模拟技术不但可以了解材料的内部微观性质及宏观力学行为,而且可以在实验前预测其力学性能,降低研发成本。分子动力学是在原子、分子水平上求解多体问题的重要模拟方法之一,可以预测纳米尺度上材料的力学特性。
单晶或多晶材料的典型晶体结构具有较为广阔的应用前景,深受研究者的青睐[3-6]。孙寅璐等[7]利用分子动力学模拟多晶面银在拉伸载荷下的变形行为,结果表明,多晶面银在低应变速率下出现脆性断裂,而在高应变速率下出现韧性断裂。刘宏西等[8]运用分子动力学模拟多晶镍在受拉过程中的塑性变形行为,结果表明,在多晶镍受拉过程中,晶界滑移诱导晶间变形,位错滑移诱导晶内变形。刘晓波等[9]采用分子动力学研究了Al2Cu拉伸变形,发现Al2Cu对温度十分敏感,温度上升使Al2Cu的原子动能成倍增加,导致塑性变强但抗拉强度明显下降。HORSTEMEYER等[10]采用了分子动力学模拟研究了单晶镍剪切性能,结果表明,在自由表面上成核的位错对原始材料的微观变形起着至关重要的作用。OGAT等[11]采用分子动力学研究氮化硅单晶剪切变形特性,发现大量滑移体系和刃型位错造成晶体突然断裂。LI等[12]采用分子动力学模拟研究单晶镍在不同温度下的剪切行为,结果表明,剪切模量和剪切模型的大小与剪切方向无关,剪切模量随温度的升高而减小。LU等[13]模拟了纳米金属层在基面上的剪切力学性能,发现随着加载温度的升高,抗拉强度明显下降。BUSTINGORRY等[14]研究了剪切速率对晶态-非晶态转变的影响,经研究发现剪切力在接近力学失稳点时可能有助于相变;稳态剪切过程中,非晶态和晶态可能共存,局部剪切变形原子重排仅局限于非晶态。
单晶/多晶复合体的结合区是单晶与多晶镍原子相互迁移而形成的扩散层。研究扩散层对单晶/多晶镍组合体微观力学性能的影响对了解宏观材料制备过程中的某些力学行为具有指导意义,因此近年来有关不同晶体的组合体研究受到人们的重视。不同应变速率和温度对结合区界面位错网结构演化影响不同,造成的位错运动和变形机制不同,从而引起结合区界面宏观力学性能也不同[15]。ZHU等[16]用分子模拟方法模拟单晶镍中γ/γ′相界面的结构动力学,结果发现,界面失配应力影响γ/γ′相界面的稳定性。JANG等[17]采用分子动力学研究碳与乙烯基酯之间的界面力学性能。发现碳表面附近聚合物的形成影响碳与乙烯基酯之间载荷的传递。张岩等[18]运用分子动力学研究Cu/Ni纳米晶薄膜的力学性能,结果表明,在塑性变形阶段依靠界面及镍原子间较强的相互作用力能够阻碍位错的传播,起到强化作用。成聪等[19]用分子动力学模拟纳米多晶Cu/Ni薄膜在不同应变率下的变形行为,结果表明,应变速率增加有利于堆垛层错的形成,较高的应变速率下具有较高的屈服极限和应变速率敏感性。张海峰等[20]利用分子动力学研究Cu/Ti复合体在单轴拉伸过程中的微观力学行为,结果发现,在拉伸载荷作用下,位错优先在Cu/Ti异质界面处形核并沿着 {111}晶面向铜层内部运动,随着位错的增殖,位错之间发生交互作用并在铜层内形成插入型层错和形变孪晶;当应变速率达到1×1011 s-1时,模型在变形过程中会出现非晶化,且随着应变增加表现出应变硬化现象。李亨等[21]采用分子动力学模拟研究两种孪晶压缩过程界面的迁移方式,结果显示, 孪晶界面上存在周期性的界面位错,阻碍了孪晶界的移动,并且位错滑移至孪晶界面处堆积,为孪晶界的迁移提供能量。
孪晶界面上存在周期性的界面位错,阻碍了孪晶界的移动,并且位错滑移至孪晶界面处堆积,为孪晶界的迁移提供能量。
目前,整体叶盘的研究大都集中在结构的加工工艺方面,鲜有整体叶盘结构在涡轮级内应用的相关研究,尤其是双晶态整体叶盘盘叶连接区工作特点的研究。在该结构得到应用之前,对其服役后可能存在的问题展开研究十分必要。为此,本工作采用分子动力学,以整体叶盘结合区单晶/多晶镍复合体作为研究对象,探讨剪切速率及温度对单晶/多晶镍复合体剪切性能的影响。
1 模型与方法
不同晶态镍分子动力学模型如图1所示。
镍的晶格常数为0.352 nm,单晶镍的晶向为X=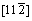 、Y=
、Y= 、Z=
、Z=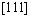 ,考虑到原子数目比较少,表面原子在总原子数中所占的比例就大,使用有限的原子数来模拟实际晶体中原子的运动,须考虑表面对体结构中原子运动的影响,为避免这种影响,所以在Y和Z方向均施加周期性边界条件。在X轴方向施加非周期性边界条件,模拟沿X轴方向剪切变形。剪切过程中采用NVT系综,牛顿运动方程用Verlet方法求解。观察剪切应力-应变曲线首次峰值确定剪切强度
,考虑到原子数目比较少,表面原子在总原子数中所占的比例就大,使用有限的原子数来模拟实际晶体中原子的运动,须考虑表面对体结构中原子运动的影响,为避免这种影响,所以在Y和Z方向均施加周期性边界条件。在X轴方向施加非周期性边界条件,模拟沿X轴方向剪切变形。剪切过程中采用NVT系综,牛顿运动方程用Verlet方法求解。观察剪切应力-应变曲线首次峰值确定剪切强度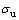 。设置温度(T)为300 K,剪切速率(v)为2
。设置温度(T)为300 K,剪切速率(v)为2  /ps分别对单晶镍、多晶镍、单晶/多晶镍复合体进行剪切模拟。
/ps分别对单晶镍、多晶镍、单晶/多晶镍复合体进行剪切模拟。
嵌入原子势(EAM)[22],是基于有效介质理论的半经验多体势函数,适合模拟原子的相互作用,可表示为
 (1)
(1)
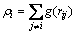 (2)
(2)
式中: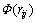 为传统对势;rij为i与j原子间的距离;
为传统对势;rij为i与j原子间的距离;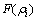 为镶嵌能;
为镶嵌能;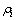 为除第i个原子以外其他所有原子j的核外电子在原子i处所产生的电子云密度(
为除第i个原子以外其他所有原子j的核外电子在原子i处所产生的电子云密度( )之和。函数的具体形式和参数可以通过ACKLAND等[23]提出的方法拟合得到。
)之和。函数的具体形式和参数可以通过ACKLAND等[23]提出的方法拟合得到。
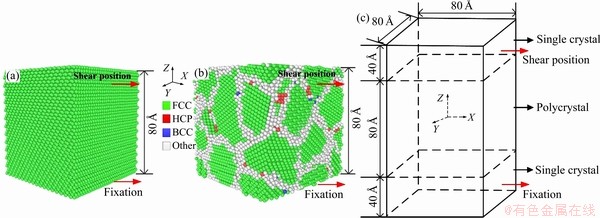
图1 不同晶态镍分子动力学模型
Fig. 1 Molecular dynamics model of single crystal Ni (a), polycrystalline Ni (b) and single crystal/polycrystalline Ni composites (c)
2 结果与讨论
2.1 晶态对晶体镍剪切性能的影响
图2(a)所示分别为不同晶态晶体镍剪切变形的应力-应变曲线,图2(b)所示为不同应变下界面处晶体镍的径向分布函数。图2(c)所示为不同应变下界面处晶体镍FCC和其他(Other)原子所占百分比。
从图2(a)中可以看出,不同晶态镍剪切在变形初始阶段应力-应变基本保持线性关系。塑性变形开始后,对于不同晶态镍的剪切变形曲线差别较大。相比于单晶镍,由于多晶镍的加入,单晶/多晶镍复合体的整体剪切应力水平有所下降,其剪切强度显著小于单晶镍的剪切强度。从图2(a)还可以看出,弹性变形阶段原子系统的原有排列结构基本保持不变。在塑性变形阶段,应力-应变曲线出现较大波动与微观变形密切相关。单晶镍中部分原子金属键断裂导致应力突然下降,而之后又出现原子迁移使得原子系统的能量升高,此时应力-应变曲线出现了新的上升,以此往复造成曲线出现波动直至降为零。与单晶镍相比,随着剪应变增大,多晶镍晶界处畸变能加大,原子迁移导致空位增多。另外,应变储存能增大,导致多晶镍晶界附近有少量的金属键断裂,塑性变形由此发生,多晶镍应力-应变曲线呈平缓状下降。如图2(b)所示,不同应变下界面处晶体镍径向分布函数呈若干尖锐峰,且随着剪应变增加,径向分布函数峰值增大。表明界面晶体镍非晶化程度较低,表现出明显的长程有序。单晶/多晶镍复合体界面系统进入塑性变形阶段,部分位错滑移出晶体。随着剪应变增加,剪切应变储存能随之增加,到达某个范围时剪切变形触发非晶相突然成核,导致部分其他无序原子转化为FCC有序原子,引起应变硬化。所以对应于图2(a)中单晶/多晶镍复合体的剪切应力-应变曲线,其整体剪切应力水平要高于多晶镍。在这种情况下,剪切变形只是帮助单晶/多晶镍复合体界面中两个相之间的转换,这与文献[6, 14]描述较为吻合。然后界面系统演化为稳态,其中非晶态原子的比例变为常数,如图2(c)所示。
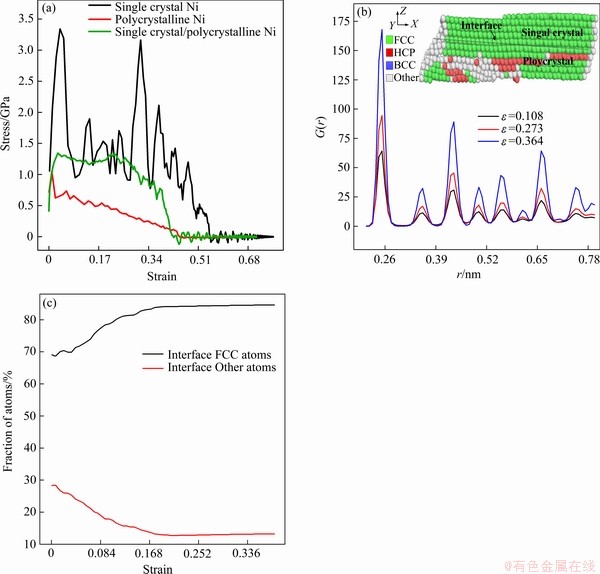
图2 不同晶态晶体镍剪切变形的应力-应变曲线,不同应变下界面处晶体镍的径向分布函数及不同应变下界面处晶体镍FCC和其他原子所占百分比
Fig. 2 Shear stress-strain curves of nickel crystals with different grain morphology(a), radial distribution function on single crystal/ polycrystalline Ni composites interface side with different strains(b), and percentages of crystalline nickel FCC and other atoms on single crystal/ polycrystalline Ni composites interface side with different strains(c)
为了更直观了解镍晶体微观变形机理,对模型做切片处理。图3所示为不同晶粒形态晶体镍的剪切原子图。如图3(a)和(b)中所示,单晶镍中堆垛层错位错网能有效地抑制混乱,引起应力强化,继续增加剪应变则产生刃型位错,进一步阻碍了变形。当剪应变达到0.524时,变形是由位错滑移主导。在塑性变形阶段形成的位错不能充分传播,并进一步滑移出晶体,如图3(c)所示。因此在应力的每一个下降阶段,位错都会发生成核、传播并最终湮灭,晶体镍恢复到FCC有序结构。
如图3(d)、(e)和(f)所示,位错滑移由横向变为切向,多晶镍与单晶镍剪切变形相比,随着应变的增加,非晶化程度明显。从图3(e)可以看出,大量刃型位错聚集,此时多晶镍晶界左右两侧原子金属键相继断裂,随着塑性变形加大导致更多的原子金属键断裂,此时刃型位错进一步聚集。如图3(f)所示,边界两侧出现空位。随着空位的增多,晶界范围进一步被压缩, 晶界变形趋于复杂化,出现了类似于宏观材料颈缩现象。
如图3(g)、(h)和(i)所示,单晶/多晶镍复合体的位错主要集中在多晶处。如图3(h)所示,塑性变形是位错滑移引起的。螺型位错沿滑移面有序过渡到刃型位错。随着剪切应变的增加,单晶/多晶镍复合体晶界进一步被压缩,空位随之增多,形成断裂区,如图3(i)所示。从图3(c)、(f)和(i)中可见,晶体镍都出现类似于宏观材料颈缩的现象,靠近颈缩区的原子发生局部错动和重排。超过屈服点后,部分FCC晶体转变为非晶态,直至材料断裂。
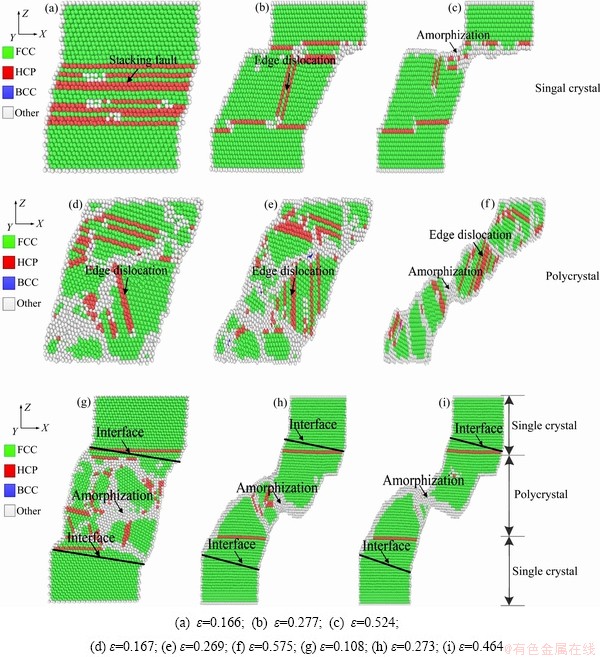
图3 不同晶态晶体镍的剪切原子图
Fig. 3 Shear deformation snapshots of nickel crystals with different crystalline state
2.2 剪切速率对单晶/多晶镍复合体剪切性能的影响
图4(a)所示为不同剪切速率下单晶/多晶镍复合体剪切应力-应变的曲线;图4(b)所示为不同剪切速率下单晶/多晶镍复合体剪切模量变化曲线,根据文献[24-26]记载取应变范围为0.5%数据拟合得到单晶/多晶镍复合体剪切模量大小。图5所示为剪切速率为3 /ps时单晶/多晶镍复合体剪切原子图。
如图4(a)所示,由于原子运动所设定的时间尺度,只能在很短的时间内进行模拟,所以这里所采用的剪切速率远高于实验。同时由于计算规模有限,造成单晶/多晶镍复合体原子驰豫后在高剪切速率下初始应力不为零。这是由于高剪切速率下表面原子失去邻近原子,形成断键,导致表面原子的配位数与内部原子不同。表面原子由于受力对称性被打破,而偏离平衡位置,产生表面张力,同时表面原子还受到内部原子的拉应力作用,这与文献[4]所述基本吻合。在本模拟中,当达到极高的剪切速率时,会出现瞬间断裂现象。在剪切速率-剪切模量曲线中,在线性弹性阶段曲线呈线性增加。在弹性区域以外,到达第一个峰值点后,单晶/多晶镍复合体出现剪切应变硬化现象。随着变形量加大,应力-应变曲线出现波动,应力出现下降,直至为零。这是由于剪切塑性变形开始后的一段时间只有少量的原子金属键断裂,主要是多晶镍晶界的变形与迁移。高剪切速率导致两个连续加载步之间的时间间隔小于晶体镍中所有原子重新达到平衡所需要的时间,使其在整个剪切过程中都难以达到平衡状态,剪切速率越大这种非平衡状态越明显。而且,晶界的存在对位错的形成和滑移运动产生阻碍作用,剪切速率越大这种阻碍作用越明显,随着切应变继续增加造成原子处于混乱状态,材料进一步变形的阻力加大,所以在这个区间剪切模量随着剪切速率的增大而增大。如图4(b)所示。但随着变形量的加大,这种稳定失衡,以及一些新的金属键相继断裂,导致应力-应变曲线产生急降。此后多晶镍处晶界附近原子迁移和重组,晶界进一步变形,单晶/多晶镍复合体又形成暂时的稳定状态,应力-应变曲线产生新的上升,如此循环,直至微型孔洞萌生后,应力迅速降为零。
如图5(a)和(b)所示,在塑性流动过程中发生应力晶化现象伴随明显的晶核形成与生长过程,随着应变的增加晶界迁移现象明显,变形是由原子迁移引起的。图5(a)中可以看出,形成的位错不能充分传播,并进一步滑移出晶体。图5(b)中显示出由于剪应力的影响,模型的位错主要集中出现在单晶镍侧,多晶镍一侧也有少量位错的出现。如图5(c)所示,靠近颈缩区的多晶镍原子排列不规则造成缺陷,在剪切变形时易在多晶镍一侧出现大量无序原子,无序原子向剪切力方向迁移,模型中部原子逐渐减少,使得颈缩区的多晶镍原子承载能力下降,空位增多,因而造成材料的断裂。所以在较高剪切速率下,单晶/多晶镍复合体变形机制为晶界迁移、位错滑移和部分非晶化。
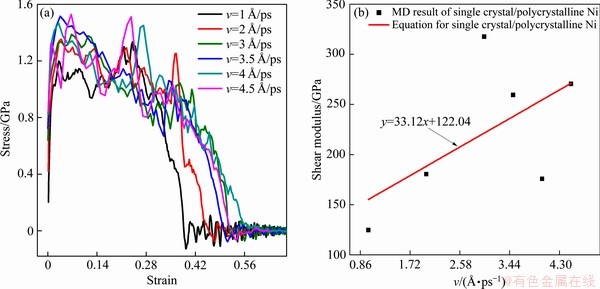
图4 不同剪切速率下单晶/多晶镍复合体剪切应力-应变的曲线及不同剪切速率下单晶/多晶镍复合体剪切模量变化曲线
Fig. 4 Shear stress-strain curves(a) of single crystal/polycrystalline Ni composites and shear modulus curves(b) of single crystal/polycrystalline Ni composites under different shear loading rates
2.3 温度对单晶/多晶镍复合体剪切性能的影响
图6(a)所示为不同温度下单晶/多晶镍复合体剪切应力-应变的曲线;图6(b)所示为单晶/多晶镍复合体剪切模量随温度的变化曲线。图6(c)所示为不同温度下单晶/多晶镍复合体剪切强度变化曲线。
如图6(a)所示,应力-应变曲线在到达第一个峰值点,随后进入塑性变形阶段,随着剪应变增加,应力-应变曲线出现波动,直至为零。在一定范围内,随着温度升高,应力-应变曲线第一次所达到的峰值逐渐下降,说明很小的剪应力可以引起较大的变形。如图6(b)所示,随着温度增加,FCC原子迅速转变为无序的非晶结构,造成单晶/多晶镍复合体承载能力迅速下降,剪切模量也随之急降。塑性阶段随着剪应变增加,试样内部尤其是多晶镍晶界附近原子及空位的迁移与重组加剧,系统能量波动变化,晶界形状不断变化,导致新的原子金属键断裂。随着越来越多的原子金属键断裂,导致材料内部原子无序化加剧。此阶段多晶Ni晶界范围逐渐缩小,晶粒间相互作用逐渐减弱,无序原子沿剪切力方向移动,试样中部原子逐渐减少,空位增多,使得颈缩区的多晶镍原子承载能力下降,因而造成了材料的断裂。由图6(c)容易看出,在温度为300 K至1300 K范围内,以1100 K作为分界线,为此引入温度阈值。当温度小于1100 K时,温度对单晶/多晶镍复合体剪切强度的影响很小;而当温度高于1100 K时,由于温度升高,单晶/多晶镍复合体非晶化程度加剧,大量原子无序化造成单晶/多晶镍复合体剪切强度急剧下降,温度越高这种现象越加明显,可以将1100 K视作阈值。
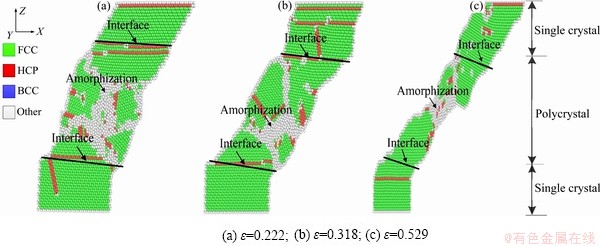
图5 剪切速率为3 /ps时单晶/多晶镍复合体剪切原子图
Fig. 5 Shear deformation snapshots of single crystal/polycrystalline Ni composites under a shear loading rate of 3 /ps
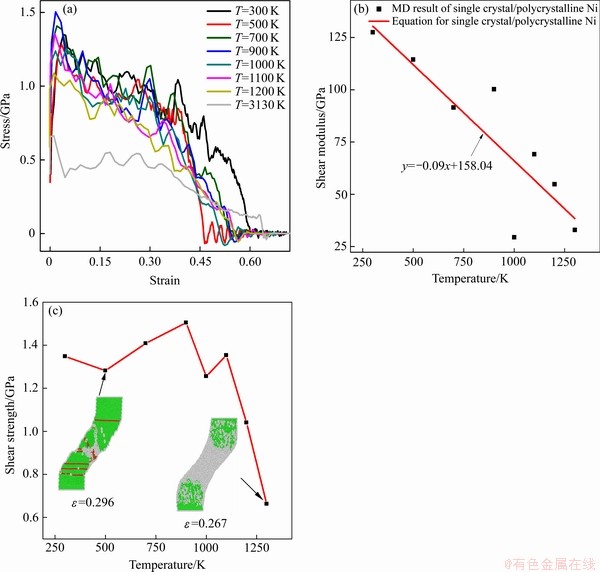
图6 不同温度下单晶/多晶镍复合体剪切应力-应变的曲线,不同温度下单晶/多晶镍复合体剪切模量曲线及不同温度下单晶/多晶镍复合体剪切强度变化曲线
Fig. 6 Shear stress-strain curves of single crystal/polycrystalline Ni composites(a), shear modulus curves of single crystal/polycrystalline Ni composites at different temperatures(b), and shear strength curves of single crystal/ polycrystalline Ni composites at different temperatures(c)
3 结论
1) 不同晶态镍剪切变形之初应力-应变基本保持线性关系。塑性变形开始后,对于不同晶态镍的剪切变形曲线差别较大。相比于单晶镍,由于多晶镍的加入,单晶/多晶镍复合体的整体剪切应力水平有所下降,其剪切强度显著小于单晶镍的剪切强度。单晶/多晶镍复合体界面系统进入塑性变形阶段,部分Other无序原子转化为FCC有序原子,引起应变硬化,所以单晶/多晶镍复合体整体剪切应力水平要高于多晶镍。不同晶态镍剪切变形进入塑性阶段后,随着塑性变形加大更多金属键断裂,在左右两侧形成裂纹。随着空位的增加,出现类似于宏观材料的颈缩现象,靠近颈缩区的原子发生局部错动和重排。
2) 随着剪切速率的增加,单晶/多晶镍复合体的剪切模量逐渐增大。在高剪切速率下,晶体镍变形机制为晶界迁移、位错滑移和部分非晶化。
3) 随着温度的增加,单晶/多晶镍复合体剪切模量呈现下降趋势。当温度超过1100 K时,单晶/多晶镍复合体剪切强度急剧下降。在温度为300 K至1300 K范围内,根据单晶/多晶镍复合体对温度的敏感程度,可以将1100 K视作阈值。
REFERENCES
[1] 刘 涛, 邓 强, 刘 源, 龙 超. 某型航空发动机整体叶盘强度分析[J]. 机械研究与应用, 2015, 28(4): 94-96.
LIU Tao, DENG Qiang, LIU Yuan, LONG Chao. Strength analysis of an aero engine blisk[J]. Mechanical Research & Application, 2015, 28(4): 94-96.
[2] 秦德胜, 陈宝延, 孙纪宁. 整体叶盘对涡轮叶盘间传热强化的数值研究[J]. 战术导弹技术, 2015, 26(2): 49-55.
QIN De-sheng, CHEN Bao-yan, SUN Ji-ning. Numerical simulation on the enhanced heat transfer effects of the blisk to the blades and disk in the turbine[J]. Tactical Missile Technology, 2015, 26(2): 49-55.
[3] CHANG L, ZHOU C Y, WEN L L, LI J, HE X H. Molecular dynamics study of strain rate effects on tensile behavior of single crystal titanium nanowire[J]. Computational Materials Science, 2017, 128: 348-358.
[4] 郑 茂. 金属单晶拉伸力学性能及缺陷行为的分子动力学模拟[D]. 南京: 南京理工大学, 2007.
ZHENG Mao. Molecular dynamics simulation of tensile mechanical properties and defect behavior of metal-single crystal[D]. Nanjing: Nanjing University of Science and Technology, 2007.
[5] 喻 娟, 张效迅, 吉建莎, 黄 丹, 习 武. 压应力条件下不同晶界对石墨烯弯曲变形的影响[J]. 中国有色金属学报, 2015, 25(12): 3452-3460.
YU Juan, ZHANG Xiao-xun, JI Jian-sha, HUANG Dan, XI Wu. Effect of grain boundary on bending deformation of graphene under compressive stresses[J]. The Chinese Journal of Nonferrous Metals, 2015, 25(12): 3452-3460.
[6] 李小凡, 胡望宇, 肖时芳, 邓辉球. 单晶和多晶钼纳米丝轴向拉伸的模拟对比[J]. 中国有色金属学报, 2009, 19(11): 102-106.
LI Xiao-fan, HU Wang-yu, XIAO Shi-fang, DENG Hui-qiu. Comparison of simulation of single-crystalline and polycrystalline Mo nanowires under uniaxial tensile strain[J]. The Chinese Journal of Nonferrous Metals, 2009, 19(11): 102-106.
[7] 孙寅璐, 刘丽娜, 文 畅, 赵健伟. 多晶面银纳米线拉伸形变的分子动力学模拟[J]. 复旦学报(自然科学版), 2018, 57(4): 94-102.
SUN Yan-lu, LIU Li-na, WEN Chang, ZHAO Jian-wei. Molecular dynamics simulation on the tensile deformation of the faceted Ag nanowire[J]. Journal of Fudan University (Natural Science), 2018, 57(4): 94-102.
[8] 刘宏西, 周剑秋. 多晶Ni纳米线拉伸变形过程尺寸效应的分子动力学模拟[J]. 南京工业大学学报(自然科学版), 2016, 38(3): 8-12.
LIU Hong-xi, ZHOU Jian-qiu. Sample size effect of polycrystalline Ni nanowires under tension: Molecular dynamics simulation[J]. Journal of Nanjing University of Technology(Natural Science Edition), 2016, 38(3): 8-12.
[9] 刘晓波, 熊 震, 方 洲, 李 艳. Al2Cu拉伸变形的分子动力学模拟[J]. 中国有色金属学报, 2018, 28(9): 1746-1754.
LIU Xiao-bo, XIONG Zheng, FANG Zhou, LI Yan. Molecular dynamics simulation of tensile deformation of Al2Cu[J]. The Chinese Journal of Nonferrous Metals, 2018, 28(9): 1746-1754.
[10] HORSTEMEYER M F, BASKES M I, PLIMPTON S J. Length scale and time scale effects on the plastic flow of fcc metals[J]. Acta Materialia, 2001, 49(20): 4363-4374.
[11] OGATA S, KITAGAWA H, HIROSAKI N, HATANAKAY, UMEZU T. Molecular dynamics simulation of shearing deformation process of silicon nitride single crystal[J]. Computational materials science, 2002, 23(1/4): 146-154.
[12] LI L, HAN M. Shear behaviors of single crystal nickel at different temperatures: Molecular dynamics simulations[J]. Applied Physics A, 2015, 119(3): 1101-1107.
[13] LU X, GUO X, YIN J, WEI Y, NAN X, DONG Q, MA Y, LA P. Shear mechanical property of β-Si3N4 nano-thin layers in the basal plane using molecular dynamics simulations[J]. Materials Science and Engineering A, 2015, 648: 72-79.
[14] BUSTINGORRY S, JAGLA E A, DOMINGUEZ D. Molecular dynamics simulation of shear in a pressure- induced amorphous model system[J]. Journal of Non-crystalline Solids, 2006, 352(28/29): 3074-3079.
[15] 吴文平. 镍基单晶高温合金的界面微结构及定向粗化行为分析[D]. 北京: 北京交通大学, 2010.
WU Wen-ping.Analysis of Interfacial Microstructure and Directional Coarsening behavior of Ni-based single crystal superalloys[D]. Beijing: Beijing Jiaotong University, 2010.
[16] ZHU T, WANG C. Misfit dislocation networks in the γ/γ′, phase interface of a Ni-based single-crystal superalloy: Molecular dynamics simulations[J]. Physical Review B, 2005, 72: 014111.
[17] JANG C, LACY T E, GWALTNEY S R, TOGHIANI H, PITTMAN Jr C U. Interfacial shear strength of cured vinyl ester resin-graphite nanoplatelet from molecular dynamics simulations[J]. Polymer, 2013, 54(13): 3282-3289.
[18] 张 岩, 肖万伸. 含Ni夹杂的纳米晶Cu基体力学性能分子动力学模拟[J]. 材料工程, 2018, 46(4): 104-110.
ZHANG Yan, XIAO Wan-shen. Molecular dynamics simulations on mechanical properties of substrate Cu with impurity Ni[J]. Journal of Materials Engineering, 2018, 46(4): 104-110.
[19] 成 聪, 陈尚达, 吴勇芝, 黄鸿翔. 不同应变率下纳米多晶Cu/Ni薄膜变形行为的分子动力学模拟[J]. 材料工程, 2015, 43(3): 60-66.
CHEN Cong, CHEN Shang-da, WU Yong-zhi, HUANG Hong-xiang. Molecular dynamics simulations of deformation behaviors for nanocrystalline Cu/Ni films under different strain rates[J]. Journal of Materials Engineering, 2015, 43(3): 60-66.
[20] 张海峰, 闫海乐, 贾 楠, 金剑锋, 赵 骧. Cu/Ti纳米层状复合体塑性变形机制的分子动力学模拟研究[J]. 金属学报, 2018, 54(9): 121-130.
ZHANG Hai-feng, YAN Hai-le, JIA Nan, JIN Jian-feng, ZHAO Xiang. Exploring plastic deformation mechanism of multilayered Cu/Ti composites by using molecular dynamics modeling[J]. Acta Metallurgica Sinica, 2018, 54(9): 121-130.
[21] 李 亨, 刘 钊, 张 真, 彭金华. 镁合金中 、
、 两种孪晶界面的可动性比较[J]. 中国有色金属学报, 2019, 29(3): 86-94.
两种孪晶界面的可动性比较[J]. 中国有色金属学报, 2019, 29(3): 86-94.
LI Heng, LIU Zhao, ZHANG Zhen, PENG Jin-hua. Mobility comparison of twin interfaces between and for magnesium alloy[J]. The Chinese Journal of Nonferrous Metals, 2019, 29(3): 86-94.
[22] MISHIN Y, FARKAS D, MEHL M J, PAPACONSTAN- TOPOULOS D A. Interatomic potentials for monoatomic metals from experimental data and ab initio calculations[J]. Physical Review B, 1999, 59(5): 3393-3407.
[23] ACKLAND G J, TICHY G I, VITEK V, FINNIS M W. Simple N-body potentials for the noble metals and nickel[J]. Philosophical Magazine A, 1987, 56(6): 735-756.
[24] WANG Z, ZU X, LI Z, GAO F. Amorphous layer coating induced brittle to ductile transition in single crystalline SiC nanowires: An atomistic simulation[J]. Journal of Physics D: Applied Physics, 2008, 41(15): 155419.
[25] LUO F, CHENG Y, CHEN X R, CAI L C, JING F Q. The melting curves and entropy of iron under high pressure[J]. Journal of Chemical & Engineering Data, 2011, 56(5): 2063-2070.
[26] LI L, HAN M. Shearing single crystal copper in molecular dynamics simulation at different temperatures[J]. Computational Materials Science, 2014, 87: 145-149.
Molecular dynamics simulation on shear mechanical properties of single crystal/polycrystalline Ni composites
LI Yuan-cai, JIANG Wu-gui, ZHOU Yu
(School of Aeronautical Manufacturing Engineering, Nanchang Hangkong University, Nanchang 330063, China)
Abstract: The integral blisk technology has been used to connect single crystal Ni turbine blades and polycrystal Ni discs with different performances into an integral blisk, but it has brought a high-risk failure problem in the joint area of the blisk, which seriously restricts the development of the technology. The shear mechanical properties of single crystal/polycrystalline Ni composites were studied by the molecular dynamics method. Firstly, the influence of crystalline state on the shear mechanical properties was discussed. Then, the effects of shear rate and temperature on the shear mechanical behavior of the single crystal/polycrystalline Ni composites were examined. The results show that, compared with single crystal Ni, the overall shear stress of single crystal/polycrystalline Ni composites decreases due to the addition of polycrystalline Ni. The shear strength of the single crystal/polycrystalline Ni composites is significantly smaller than that of single crystal Ni. After the shear deformation of the different crystalline Ni enters the plastic state, a phenomenon similar to that of macroscopic material necking occurs, and the atoms near the necking zone are locally dislocated, rearranged, and partially amorphized. The shear modulus of the single crystal/polycrystalline Ni composites increase with the increase of shear loading rate, while they shows a downward trend with temperature rising.
Key words: integral blisk; single crystal/polycrystalline Ni composites; molecular dynamics; shear rate effect; temperature effect
Foundation item: Project(11772145) supported by the National Natural Science Foundation of China
Received date: 2019-08-14; Accepted date: 2019-12-13
Corresponding author: JIANG Wu-gui; Tel: +86-791-83863735; E-mail: jiangwugui@nchu.edu.cn
(编辑 何学锋)
基金项目:国家自然科学基金资助项目(11772145)
收稿日期:2019-08-14;修订日期:2019-12-13
通信作者:江五贵;教授,博士;电话:0791-83863735;E-mail:jiangwugui@nchu.edu.cn

