文章编号: 1004-0609(2006)07-1171-06
铌和钨(001)面熔化的分子动力学模拟
阳喜元1, 胡望宇2, 袁晓俭2
(1. 湖南文理学院 物理与电子科学系, 常德 415000;
2. 湖南大学 应用物理系, 长沙 410082)
摘 要: 采用改进分析型嵌入原子方法(MAEAM), 对铌、 钨晶体的(001)面预熔和熔化过程进行分子动力学模拟。 通过计算不同温度的原子位置、 层原子密度、 层结构因子、 径向分布函数等物理量, 研究(001)面的微观结构随温度的变化。 结果表明, (001)面在低于其完整晶体的熔化温度出现预熔。 另外, 通过将该面预熔摩尔分数对温度进行拟合发现, 可以近似地预测(001)面的预熔过程。
关键词: 铌; 钨; 熔化; 预熔; 改进分析型嵌入原子法(MAEAM); 分子动力学(MD)
中图分类号: TG146.4 文献标识码: A
Molecular dynamics simulations of
melting behavior of (001) plane of niobium and tungsten
YANG Xi-yuan1, HU Wang-yu2, YUAN Xiao-jian2
(1. Department of Physics and Electronics, Hunan University of Arts and Science,
Changde 415000, China;
2. Department of Applied Physics, Hunan University, Changsha 410082, China)
Abstract: Using the modified analytic embedded-atom method (MAEAM), the molecular dynamics (MD) simulation was performed to study the premelting and melting behavior of (001) plane of niobium and tungsten. The physical properties including the atomic positions, the layer atomic density and the layer structure factor were calculated to disclose the relations between their microstructures and temperature. The results indicate that there are some quasiliquid layers on plane (001) at some temperature below the melting points of the prefect crystals. In addition, the premelting mole fraction of the surface of these two metals is fitted with the corresponding temperature to obtain a relational expression which is able to forecast the disordering progress of this surface.
Key words: niobium; tungsten; melting; premelting; modified analytic embedded-atom method (MAEAM); molecular dynamics (MD)
晶体熔化是一个重要的相变过程, 表面对晶体的熔化影响很大[1-8]。 而铌、 钨具有高熔点等特征, 在高温下应用广泛, 考虑到原子振动非谐效应对其结构和性能的影响, 对这两种金属的预熔和熔化现象进行深入研究有重要意义。 目前, 由于受条件的限制, 对其结构和性能随温度变化关系的研究主要集中在低温领域[9, 10], 而高温状态下的研究涉及甚少, 且研究所得的结果也不尽令人满意, 其原因是铌、 钨具有体心立方结构特征, 其第一、 二近邻原子相互作用等因素的影响明显[11], 在高温条件下, 其结构特征和性能随温度变化难以确定。 因此, 开展对铌、 钨表面预熔和熔化的研究有重要意义。 本文作者采用改进分析型嵌入原子势函数, 用分子动力学方法[12]研究铌、 钨的(001)面微观结构随温度的变化, 深入分析该面熔化过程。
1 研究方法和势函数
1.1 研究方法
本文采用等温等容系综(NVT), 在零压条件下运用MD方法模拟Nb、 W的(001)面预熔和熔化。 构建(001)面包含(5a×7a×18a)单胞(a为晶格常数), 并在(001)面的X[100]、 Y[010]方向施加周期性条件消除边界效应。 分子动力学时间步长取0.5fs, 在低于3000K和高于此温度下分别进行0.1ns和0.5ns弛豫, 使体系平衡。 在模拟过程中, 采用速度温标调温技术[13]保持恒温。 对于层原子密度、 层结构因子、 径向分布函数等物理量的计算方法参见有关文献[14]。
1.2 势函数选取
本文采用Daw和Baskes首先提出的[15], 经张邦维、 胡望宇等[16]改进的分析型嵌入原子势(MAEAM)描述原子之间的相互作用, 其总能量的表达式为

2 结果与讨论
2.1 原子密度函数
本文所研究的密度函数取沿(001)面法线方向的原子分布为z轴坐标, 如图1所示。 在一定温度下(Nb、 W分别为1900、 2700K), 其表面附近出现无序, 原因是表面的一部分原子成为自由原子, 并吸附在表面而形成准液态层, 说明在一定温度下, (001)面出现预熔, 与文献[5]结论一致。 随着温度升高, 表面两层原子密度函数分布曲线的峰值降低、 变宽, 而峰谷也已升高、 变窄, 其准液态层不断向内层原子扩展而变厚。 表面处密度函数曲线略微向上, 说明内层原子脱离平衡位置后向表层区
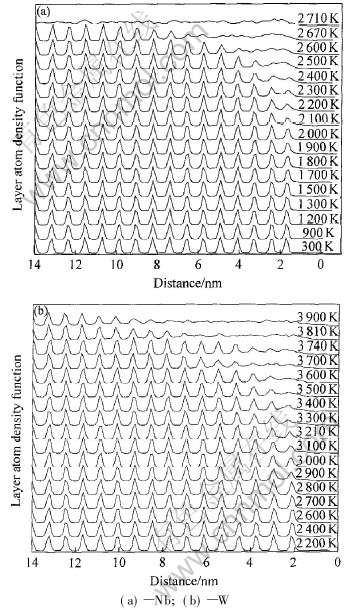
图1 铌和钨(100)面的原子密度分布函数
Fig.1 Atomic density function of plane (001) of Nb and W
域迁移。 最后, (001)面的层原子密度函数曲线已经近似于一条直线, 说明整个试样处于无序状态中, 该面熔化, 由此估算铌熔点是(2690±10)K, 低于完整晶体的熔点, 说明表面影响非常明显; 而钨的熔点为3900K左右, 略高于其完整晶体的熔点, 有待于进一步研究。
2.2 层结构因子
图2和3所示为Nb, W(001)面在不同温度下的层结构因子随其原子层数变化。 在各个温度下, 最外层原子的结构因子都比晶体内层的要小, 且随温度升高层结构因子变化显著, 与文献[3-6, 8]结果类似, 说明表面的影响很大, 其原因是由于原子的晶格振动所造成的。 当该面处于一定温度下(铌为1700K, 钨为2900K), 表层结构因子已经小于0.5, 说明表面处于无序状态, 并形成空位-吸附原子对, 预熔现象产生, 其预熔温度接近于本文由密度函数计算的结果。 随着温度升高, 各层面的层结构因子越来越小, 准液态层也由表层向晶体内层一层一层地延伸。 到2710K, 铌(001) 面层结构因子都处于水平线以下而完全处于无序状态, 低于Nb的完整晶体的熔点; 而W(001)面在3810K温度以上, 其整个薄层的层结构因子已经小于0.5, 略高于完整晶体的熔点, 与密度函数的结果一致。 同时, 随着温度升高, 相邻两温度层结构因子的差越来越大, 说明非谐效应对原子振动的影响超过了原子之间的束缚力而成为自由原子, 造成层结构因子显著减小。

图2 Nb(001)面的层结构因子随温度的变化
Fig.2 Effects of temperature on layer structure factors of plane (001) of Nb
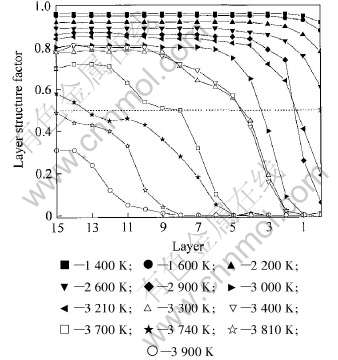
图3 W(001)面的层结构因子随温度的变化
Fig.3 Effects of temperature on layer structure factors of plane (001) of W
2.3 径向分布函数
(001)面预熔和熔化现象也可以用径向分布函数(RDF)曲线来加以解释, 如图4所示。 在一定温度下(铌在1700K左右, 钨在2900K左右), (001)面最外层原子的RDF曲线变得光滑, 最外两表层结构已经处于无序状态并形成一准液态层, 出现预熔, 文献[3-5]也有类似结果。 随着温度升高, (001)表面层的RDF曲线尖峰不断宽化, 但仍然保持其尖峰的基本形状, 说明(001)面的原子非谐效应随温度升高而增强。 但是准液态层并没有随温度升高而向里扩展, 直到接近于铌、 钨的各自熔点时, 靠近准液态层的原子出现混乱, 熔层向晶体内部扩展。 由径向分布函数所得预熔和熔化温度与层原子密度和结构因子的一致。
由于表面预熔是逐层过渡, 本文通过RDF近似确定发生预熔的原子数, 并将预熔原子分数定义为预熔原子数与总原子数比。 将预熔原子分数的自然对数与温度倒数进行数值拟合, 研究[CM(22]Nb(001)、 W(001)预熔原子分数对数随温度的变
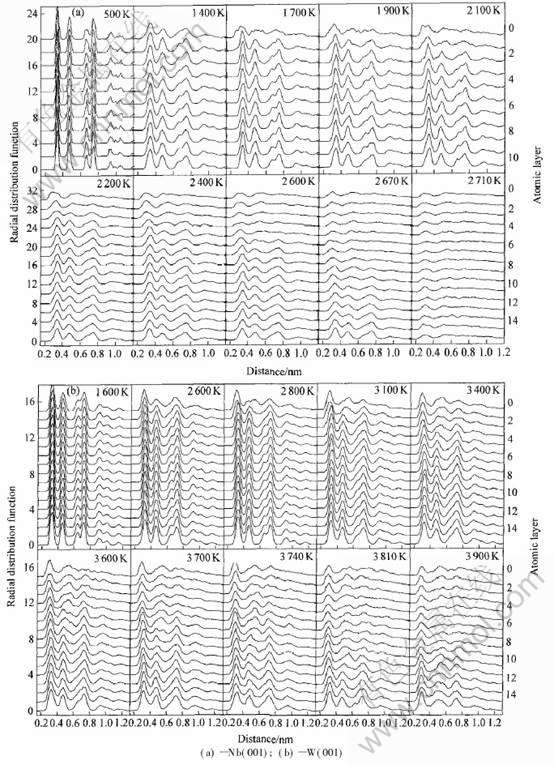
图4 Nb和W的(001)面的径向分布函数
Fig.4 Radial distribution functions of plane (001) of Nb and W
化。 图5所示为拟合曲线, 在预熔到熔点温度范围(T≤Tm), 拟合后发现铌预熔的原子分数f随温度变化可由下列函数关系式描述:

从图5的拟合情况来看, (001)面预熔原子分数随温度变化可以由式(8)加以预测, 有利于进一步研究表面的熔化进程。 对于Nb(001)面, 由预熔原子分数拟合得到的激活能等于0.89eV, 与文献[17]Nb(001)的值0.99eV接近; W(001)预熔激活能为2.2140eV, 还没有找到相关文献研究, 但大于铌(001), 说明钨的预熔和熔化温度高于Nb的。 通过表面原子预熔激活能, 可以判断不同元素相同表面预熔和熔化情况, 预熔激活能越大, 表面出现预熔现象的温度也高, 其熔点也要高。
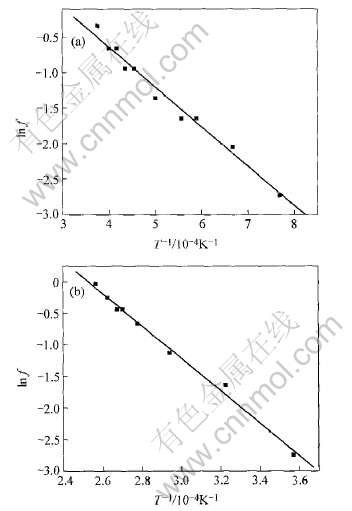
图5 Nb和W的(001)原子预熔分数对数随温度的变化
Fig.5 Effects of temperature on premelting atomic fraction of plane (001) of Nb and W
3 结论
1) 通过对(001)面的分子动力学模拟研究, 表明表面在较低温度下出现预熔, 熔化温度低于或接近完整晶体。
2) 表面作为一类缺陷, 对晶体的结构和性能有着重要的影响, 随着温度的升高, 在表面区域首先形成熔化核, 并向周围扩展。
3) 通过对预熔原子分数与温度进行拟合, 可以定性预测表面预熔随温度变化的关系。
REFERENCES
[1]Lo W S, Chen T S, Fang B S, et al. Photoelectron-diffraction studies of Nb(001)[J]. Surface Review and Letters, 1998, 5(5): 1035-1041.
[2]Frenken J W M, van der Veen J F. Observation of surface melting[J]. Phys Rev Lett, 1985, 54(2): 134-137.
[3]Lutsko J F, Wolf D, Phillpot S R, et al. Molecular-dynamics study of lattice-defect-nucleated melting in metals using an embedded-atom-method potential[J]. Phys Rev B, 1989, 40(5): 2841-2855.
[4]王海龙, 王秀喜, 梁海弋. 应变效应对金属Cu表面熔化影响的分子动力学模拟[J].物理学报, 2005, 54(10): 4836-4841.
WANG Hai-long, WANG Xiu-xi, LIANG Hai-yi. Molecular dynamics simulation of strain effects on surface melting for metal Cu[J]. Acta Phys Sin, 2005, 54(10): 4836-4841.
[5]Chen E T, Barnett R N, LandmanU. Surface melting of Ni(110)[J]. Phys Rev B, 1990, 41(1): 439-450.
[6]王海龙, 王秀喜, 梁海弋. 金属Cu体熔化与表面熔化行为的分子动力学模拟与分析[J]. 金属学报, 2005, 41(6): 568-572.
WANG Hai-long, WANG Xiu-xi, LIANG Hai-yi. Molecular dynamics simulation and analysis of bulk and surface melting processes for metal Cu[J]. Acta Metallurgica Sin, 2005, 41(6): 568-572.
[7]Foiles S M, Adams J B. Thermodyna-mic properties of fcc transition metals as calculated with the embedded-atom method[J].Phys Rev B, 1989, 40(9): 5909-5915.
[8]Kojima R, Susa M. Surface melting of copper with (100), (110), and (111) orientations in terms of molecular dynamics simulation[J]. High Temp-high Press, 2002, 34(6): 639-648.
[9]Fu C L, Freeman A J. Multilayer reconstruction and vibrational properties of W(001)[J]. Phys Rev B, 1988, 37(5): 2685-2688.
[10]Yu R, Krakauer H. Equilibrium geometry and electronic structure of the low-temperatureW(001)surface[J]. Phys Rev B, 1992, 45(15): 8671-8679.
[11]Sorkin V, Polturak E, Adlar J. Molecular dynamics study of the bcc metal vanadium(Ⅰ): Mechanical melting[J]. Phys Rev B, 2003, 68(17): 174102-1-174102-7.
[12]Nose S. A unified formulation of the constant temperature molecular dynamics methods[J]. Chem Phys, 1984, 81(1): 511-519.
[13]文玉华, 朱如曾, 周富信, 等. 分子动力学模拟的主要技术[J]. 力学进展, 2003, 33(1): 65-73.
WEN Yu-hua, ZHU Ru-zeng, ZHOU Fu-xin, et al. An overview on molecular dynamics simulation[J]. Advances in Mechanics, 2003, 33(1): 65-73.
[14]阳喜元. 难熔金属晶体(钒、 铌、 钨)低指数表面熔化的分子动力学模拟研究[D]. 长沙: 湖南大学, 2005.
YANG Xi-yuan. The molecular simulation study of the low surfaces thermodynamic melting of refractory metals(V, Nb, W)[D]. Changsha: Hunan University, 2005.
[15]Daw M S, Baskes M I. Embedded atom method: Derivation and application to impurities, surface, and other defects in metals[J]. Phys Rev B, 1984, 29(12): 6443-6453.
[16]张邦维, 胡望宇, 舒小林. 嵌入原子方法理论及其在材料科学中的应用[M]. 长沙: 湖南大学出版社, 2003. 245-246.
ZHANG Bang-wei, HU Wang-yu, SHU Xiao-lin. Theory of embedded atom method and its application to materials science―Atomic scale materials design theory[J]. Changsha: Hunan University Press, 2003. 245-246.
[17]Lekka C E, Mehl M J, Bernstein N, et al. Tight-binding simulations of Nb surfaces and surface defects[J]. Phys Rev B, 2003, 68(3): 035422-1-035422-8.
(编辑陈爱华)
基金项目: 国家自然科学基金资助项目(50371026)
收稿日期: 2005-12-21; 修订日期: 2006-03-13
通讯作者: 阳喜元, 讲师; 电话: 0736-7186121; E-mail: oyxy889@163.com

