�������þ�Ͻ���������Ĥ����Ӱ����о���չ
��Դ�ڿ����й���ɫ����ѧ��2006���11��
�������ߣ����ٷ� ������ ��С�� ������ ������ ������
����ҳ�룺1829 - 1837
�ؼ��ʣ�þ�Ͻ� ���������� �������������
Key words��magnesium alloys; anodization; electric parameters; property
ժ Ҫ���������������þ�Ͻ���ʴ�Ե�һ����Ч������ ѡ��ĵ����������Ĥ����Ӱ��ܴ� �����˵������Ƶ�ʡ� ռ�ձȡ� �����ܶȡ� �յ�ѹ�ȵĸ�� �Ƶ���ռ�ձ�������ܶȵĹ�ϵ�� �������˵��������������Ĥ����Ӱ����о���չ�� ���ŵ��Ӽ�����������Դ�ϵĹ㷺Ӧ�ã� �����豸�ķ�չ����Ϊ�������ܻ��Լ�Ƶ��Խ��Խ�ߡ�
Abstract: Anodizing is an effective method to improve the corrosion resistance of magnesium alloys, and the selected electric parameters have great effects on the properties of anodic coatings. Concepts of some electric parameters such as frequency, duty cycle, current density and final voltage were introduced and the relation between duty cycle and current density was deduced. In addition, the research progress about the effects of electric parameters on coating properties was reviewed. With the wide use of electric technology on anodizing power supply, the development trend of anodizing equipment is the intelligence and higher and higher frequency.
���ٷ�1, 2, ������1, ��С��3, ������1, ������2, ������2
(1. �����Ƽ�ʦ��ѧԺ ����ʡ���ϱ��湤���ص�ʵ����, �ϲ� 330013;
2. �й���ѧԺ�����о��� ���ϻ�����ʴ�����о�����, ���� 110016;
3. �����Ƽ�ʦ��ѧԺ ����ϵ, �ϲ� 330013)
ժ Ҫ: �������������þ�Ͻ���ʴ�Ե�һ����Ч����, ѡ��ĵ����������Ĥ����Ӱ��ܴ� �����˵������Ƶ�ʡ� ռ�ձȡ� �����ܶȡ� �յ�ѹ�ȵĸ���, �Ƶ���ռ�ձ�������ܶȵĹ�ϵ, �������˵��������������Ĥ����Ӱ����о���չ�� ���ŵ��Ӽ�����������Դ�ϵĹ㷺Ӧ��, �����豸�ķ�չ����Ϊ�������ܻ��Լ�Ƶ��Խ��Խ�ߡ�
�ؼ���: þ�Ͻ�; ��������; �����; ���� ��ͼ�����: TG174
���ױ�ʶ��: A
ZHANG Rong-fa1, 2, LI Ming-sheng1, LONG Xiao-li3,HE Xiang-ming1, SHAN Da-yong2, HAN En-hou2
(1. Jiangxi Key Laboratory of Surface Engineering,Jiangxi Science and Technology Normal University, Nanchang 330013, China;
2. Environmental Corrosion Center, Institute of Metal Research,Chinese Academy of Sciences, Shenyang 110016, China;
3. Department of Physics, Jiangxi Science and Technology Normal University,Nanchang 330013, China)
Abstract: Anodizing is an effective method to improve the corrosion resistance of magnesium alloys, and the selected electric parameters have great effects on the properties of anodic coatings. Concepts of some electric parameters such as frequency, duty cycle, current density and final voltage were introduced and the relation between duty cycle and current density was deduced. In addition, the research progress about the effects of electric parameters on coating properties was reviewed. With the wide use of electric technology on anodizing power supply, the development trend of anodizing equipment is the intelligence and higher and higher frequency.
Key words: magnesium alloys; anodization; electric parameters; property
þ�Ͻ�����ܶ�С, ��ǿ�ȡ� �ȸնȸ�, ��������ԡ� �����Ժ��Լ������������ӹ���������, �ں��ա� ������3C��Ʒ������кܴ��Ӧ��DZ��[1-4]�� ��������ʴ�Խϲ�, ��Լ��þ�Ͻ�Ĺ㷺Ӧ�á� þ�Ͻ��õķ��������е��/��ѧ�ơ� ��ѧת��Ĥ�� �л���Ϳ�㡢 ���������Լ���������������֮�Ϸ�չ�������������� ����������Ȼ�豸Ͷ�ʴ�, ����õ���������Ʒ��ʴ�Բ�����ǰ3�ַ�����, ������Һ��ʹ��������, �����ǿ���ʹ�û�������ɫ���Һ, ����ҵ����㷺ʹ�õ�þ�Ͻ���洦������[5], �������߶Դ˱��洦�������ѽ���������[6]��
Ӱ��þ�Ͻ�����������ĤЧ�������ذ���: ���Һ����ɼ���Ũ�ȡ� �����(��ѹ�� ����)���͡� ��ֵ������Ʒ�ʽ�� ��Һ�¶ȡ� pHֵ������ʱ���[7]�� ��þ�Ͻ���������������, �豸�����������dz���Ҫ�� ����, ��Ӱ������Ĥ�ijɷ֡� �о��������, ���Һ�е������ӽ�������Ĥ�дӶ�Ӱ��Ĥ������, �������ͨ���ı�����������Һ�еĴ��ʹ��̴Ӷ�������Ĥ�ijɷֲ���Ӱ�졣 �ܽ�������Ĥ�е�������, ��������ͨ�����ʵ�������/���Һ���档 ���ʱ, ������������Ǩ����3�ַ�ʽ[8]: ����Һ������缫�������ɢ, Ũ����²���������Һ�����Լ���Ǩ�ơ� ���Ƶ�, ��������ʱ, ��Һ�е�������Ҳ��������3�ַ�ʽ�� ��Һ�е������ӵ�������/���Һ�����, ���������þ���Ӻ������ӵ�Ũ�ȳ˻��Ƿ�������ܵ���ʵ��ܶȻ����ܶȻ�����, �Ⱥ���þ�Ͻ�������ɻ���� ���ڸ��������ӵ�Ũ�Ȳ�ͬ, ������Һ�е��ƶ��ٶȲ�ͬ, �����ͨ���ı������ӵĴ��ʹ��̴Ӷ�Ӱ��������������/���Һ�����Ũ��, ����Ӱ������Ĥ�ɷ֡�
���, �����������Ĥ������ò�ͽṹ�йء� þ�Ͻ�ĵ�������������Ϊ3����[9]: ��ͳ����������(traditional anodizing)�� ������(micro-arc anodizing)�ͻ�������(arcing)�� �������ij���, ��ѹ���ߵ��ٶȺܿ�, �����������ɺܱ�һ���ԵĤ, ͬʱ����������, ���ǵ�һ��, ����ͨ���������Ρ� ����ѹ����ijһ�ٽ�ֵʱ, ��Ʒ��������ܼ��� ϸС�Ļ�, ���ǵڶ���, ���������Ρ� �ڴ˽�, ��ʹ�����������������ʱ, �����������������, �γ�˲����¸�ѹ(p��102GPa, T��2��104K)[10], ʹ����Ĥ������״, ��ʱ��������Ĥ�е����������緢�� ͬʱ�ڸ���������, ����Ĥ�Ľṹ�����仯, �ɷǾ�̬��̬ת��; ����������ĶϿ�ʱ�����ʱ, �������廡��ʧ, ���ڵķŵ�ͨ���ų����ڵ��Һ�ġ���㡱�����¿�������, �ڲ��ϱ����γɶ�ṹ�� ���ڵ����, �������Ըı���������, Ҳ���Ǹı���Ʒ�ı����¶�, �Ӷ�ʹ����Ĥ�Ľṹ�����仯, ���ҿ��Ըı䡰�緢���͡���㡱ʱ��, �Ӷ��ı�����Ĥ�ı�����ò��
���, ����������Ч���йء� ʹ��ǡ���ĵ�������, �������Ч��, ������������[11], ������Դ�����ѷ��Ľ���dz���Ҫ��
�����������õ�Դ��ֱ��(DC)�� ����(AC)������֮��, ������Ҫʹ��ֱ��, Ȼ�����ֱ���ϵ��ӽ������������[12]; ��������Ŀ������к�ѹ�ͺ���֮��
1 ���������ͺ�ѹ�����ıȽ�
����ں�������[4, 9, 13-27], ��ѹ�����о��ý���[13, 28-31]�� �����������DZ�֤���������㶨, ������������, ������ѹ��������, ֱ���趨ֵ; ��ѹ�������ǽ��㶨��ѹʩ�ӵ�����, ��ʼ�����ܴ�, �ڴ�����ܶ�����������ĤѸ�������� ʹ�ú�ѹ����ʱ, ���������������Ƿdz��õĵ���, ��������ȵ�����, �����ܶȲ����½�, ���ɵ�������Խ��Խ����ĥ; �����������ɱ�֤Ĥ�����ʾ���, ����ʡʱ[32]�� ������[33]����ѹ���������о������������������о�������бȽ�, ���ֺ�ѹ������������ȱ��: 1) Ĥ�������ٶ�ʮ�ֻ���; 2) ��Ĥ���̷Ǿ��Ƚ���, ���Խ�����ѧ�����ʱ�����; 3) �ڳ�Ĥ���ڴ��ֵ�ĵ����������豸��ɺܴ�ij���� ���֮��, ��������������������Щȱ��, ��˱��ֳ�����Խ�ԡ� ���������ڴ˽��ܺ�������������
2 ֱ��������������������ĶԱ�
���ں�������, ������������ĵ���������ֱ��(DC)�� ����(AC)(��ͼ1)������3��, �������ְ�����(��)�����˫�������ַ�ʽ, ��ͼ2��
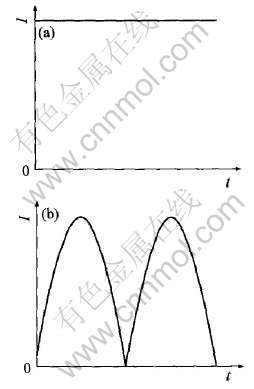
ͼ1 ֱ��(a)�ͽ���(b)����ʾ��ͼ
Fig.1 Sketch diagrams of direct current (a) and alternating current (b)
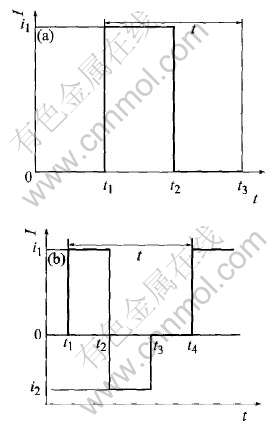
ͼ2 ��(��)�������(a)��˫�������(b)����ʾ��ͼ
Fig.2 Sketch diagrams of positive (unipolar) pulse current (a) and bipolar pulse current (b)
����ֱ���������������������Ĥ��Ӱ��, �����в�ͬ�Ŀ����� ������Ϊ����þ���渽��ǡ�����������������ӱ����dz���Ҫ, ���ǵõ��㹻�ȶ������ܵı���������Ĥ��Ψһ������ ʹ��ֱ����������, �Ʊ���������������������ر���OH-�ĸ���(OH-���ƶ��ٶȺܿ�), �Ӷ���������Ĥ���ȶ��Խϲ��Mg(OH)2�γ�; ��ʹ��һ��Ƶ�ʵĽ���������������������������������õ�ǡ���ĸ��������ӱ���, �Ӷ��γɸ���þ�Ļ�����������þ�� ����þ��, ���ʹ�����������ֱ��������Խ[34-36]; Ҳ������Ϊ���ֵ����������Ĥ�γɹ���û������Ӱ��[37]��
��������˵, ����˫����(��ͬʱ������������������ܺ�����), ����ʹ����Ĥ������, ��϶�ʸ���, ���������ڱ���ij��˾������������Դ��ȷʵ�õ������ֽ���� ������Ϊ: ����ӵ�ѹ���ڻ�����ѹ��, �ŵ�ʹ��������ֲ��¶Ⱥܸ�, ����ֱ������, ��ʱ������һֱ�л��γ�, ��������������ʱ�����Һ����, �ŵ������¶�Ѹ������, ����̬�������������緢���γɽϴ�����; ����˫����, �ڵ����ĸ�����ʱ, ������������Ϊ����, ��Һ�е�H+������Ǩ�Ʋ�����������, ���ܴ����������������, ����ʹ������/���Һ������õ���ȴ, ���Ĥ�Ŀ�϶�ʸ���, ������ò������[15]��
��������һ�ҹ�˾��������������Դ��, ��������[38]Ҳ�о���˫�����������Ʒ��ʴ�Ե�Ӱ�졣 ʵ��ʱֻҪ����˫����, ���ܸ������ܶ��趨�����С, ������ѹ����һ��ֵʱ, ��Ʒ����Ļߴ����Ա�ͬ��������µĵ������, ������, ��������Ĥ�Ŀ�϶�ʸ���, ������Ʒ����ʴ�Ա��, ���������о��ߵĽ��һ��[39]�� �������������ԭ��, �����ǻ���������̬��ת���븺��������ع����ڵ���������֮��Ľ��������������ɢ��������ϵ[39]��
�����������, �����м��������, ����ֱ���н��ܡ�
3 ������Ľ���
�������Ƶ�ʡ� �����ܶȡ� ռ�ձ�(duty cycle)���յ�ѹ(final voltage)�ȡ� ��Ϊ������������������֮��, ��˹��ղ��������ֿɷ�Ϊ: Ƶ�ʡ� �������ܶȡ� �������ܶȡ� ����ѹ�� ����ѹ�� ��ռ�ձȺ�ռ�ձȡ�
���ں������� ������IJ���, ���������ķ�������, ����6���������綨: Ƶ�ʡ� ���������i1, ���������ʱ��t2-t1, ���������i2, ���������ʱ��t3-t2�ͶϿ�ʱ��t4-t3(��ͼ2(b))��
Ƶ���ǵ�λʱ�������Ĵ���, ��һ����������ʱ��ĵ���:
![]()
![]()
�����ܶ�����Ҫ�ĵ�Դ���Ʋ���֮һ, ����ʩ������Ʒ��λ����ϵĵ���ֵ��
��ƽ������im1��ƽ������im2�ֱ�Ϊ[8]

��(��)ռ�ձȾ���һ��������������(��)��������ʱ����һ������ʱ��ı�ֵ, ��

ʽ�� ��1�ͦ�2�ֱ�Ϊ������ռ�ձȺ�����ռ�ձȡ�
��ʽ(5)��(6)����ʽ(3)��(4), �õ�

��ʽ(7)��(8)��֪, ��ƽ������im1��im2һ��, Ҳ���Ǻ�������ʱ, ռ�ձ�Խ���������ԽС��
ʵ����, �����豸��ԭ��, ͼ2(b)�е����������i1���������i2�������Ǻ㶨�����, ���Dz�����, ֻ��ƽ�������㶨�� ���ನ�ο���������8���������綨: Ƶ��, ������������ip1, �������˲ʱ����ֵi1, ���������ʱ��t2-t1, ����������ip2, �������˲ʱ����ֵi2, ���������ʱ��t3-t2�ͶϿ�ʱ��t4-t3(��ͼ3)��
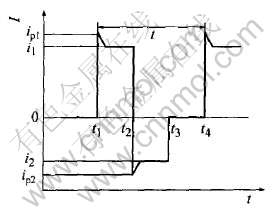
ͼ3 ʵ�ʷ�����������ʾ��ͼ
Fig.3 Sketch diagram of practical square current waveform
��t2-t1ʱ���, ����������Dz��ϱ仯��, ������ƽ��ֵ![]() �������С,
�������С,

����һ��������, ��ƽ�������ܶ�im1Ϊ

��ʽ(11)���Եó�, ���ں�������, Ҳ����һ�������еĵ���Ϊ�̶�ֵ, ��im1�̶�, ��ʱռ�ձȺ�������ʱ��ε�ƽ�������ܶ�![]() �ɷ��ȡ�
�ɷ��ȡ�
ͬ��, ������ʱ��ε�ƽ������i2��һ�������еĸ�ƽ������im2�ֱ�Ϊ

��ʽ(6)�� (12)��(13), ��
![]()
����������ķ������廹��ʵ�ʵķ�������, ��t2-t1ʱ���, ������������; ��t3-t2ʱ���, �����ļ��Է����ı�, �������������; t4-t3ʱ���Ϊ�Ͽ�ʱ�䡣 ��t3-t2��t4-t3ʱ���, ����̬������Ĥ�������̡�
�յ�ѹ����������ʱʩ������Ʒ�ϵĵ�ѹֵ�� ���������������õĵ�Դ�� ���Һ�Ļ�����ѹ�Լ�����Ĥ�������йء�
��������ʹ�ò�ͬ�������豸, ��ͬ���ĵ��Һ������ͬ����þ�Ͻ�, Ϊ��ʹ����Ĥ�ĺ��һ��, ʹ��Ƶ�ʸߵ�������Դѡ�����յ�ѹ��Ƶ�ʺܵ͵ĵ�Դ�ߡ�
���, �յ�ѹ������Һ�Ļ�����ѹ�йء� ���������ŵ��γ�����Ĥ�Ĺ���, �յ�ѹһ����ڻ�����ѹ; �෴, ���ڲ��������ŵ��γ�����Ĥ�Ĺ���, ��һ��С�ڻ�����ѹ�� ������ѹUB(breakdown or sparking voltages)�Dz�����������һ��������������ʱ, ͨ��һ̨��չ�ʽ���ؼƼ��µĵ�ѹֵ[40]�� ��������ѹ�ﵽ������ѹ��, ��ѹ�����ٶ����Խ���, ͬʱ�����ϲ����������������� �IJ���, �������ӵ�������������Ĥ���Ҵ�ʱ��������������������������(tunneling mechanism)ת���ɰ���ѩ����ֳ(avalanche multiplications)���ڵ�һ�ֻ���[41], ����Ĥ�ɷǾ�̬��ɾ��塣 ������ѹ��������[41-42]�� ���Һ�������Ũ��[42-43]�Լ����Һ�ĵ�����(��)�й�[40, 44], ���鹫ʽΪ[40]
![]()
ʽ�� aB�� bB��һ���Ľ����͵��Һ���Ϊ����, ����������������ܶ�[43-44]�� ��Һ�¶�[40-41]�� ���Һ�Ľ����ٶ�[42]�� �Ƿ��γɹ�����Ĥ�ȶ���������Ӱ����[42, 45]�� ����������о��ó�������ѹ������ܶ��й�, ����ʹ�õĵ����ܶ�Խ��, ������ѹԽ��[9]��
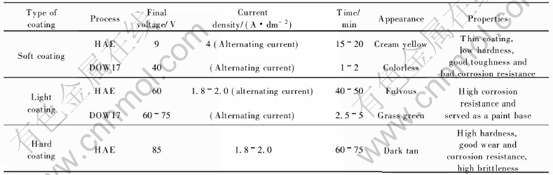
���, �յ�ѹ��������Ĥ�������йء� ����þ�Ͻ����������ľ��乤��HAE��DOW17, ���յ�ѹ�IJ�ͬ, ���Եõ�3�����ܸ����Ĥ��, ����1��
��1 HAE��DOW17���ַ�����õ�Ĥ�����ͼ�������[46]
Table 1 Coating types and properties obtained by HAE and DOW17 methods[46]
4 �������Ӱ��
4.1 Ƶ��
Ƶ�ʵĸߵ��Ǻ���������Դ�����û���һ����Ҫָ��, ����������������Դ�������������, ��Ҫ������Ƶ��Խ��Խ���Լ���������Ƽ������ⷽ��õ��˹㷺Ӧ�á�
����Ƶ�ʶ�����Ĥ��Ӱ��, Ŀǰ�о��������һ��, ������Ƶ�ʵ�����, �մɲ�����ܶ��������[47], ��ʴ�Գ��������ӵ�����[48], ����Ƶ�ʶ�����Ĥ���������洦�� ���ں�������, ����������ʱ����յ�ѹΪʵ����������о�, ����Ĥ�ĺ����Ҫ���յ�ѹ����, ��Ƶ�ʹ�ϵ���� ��������ʱ, һ��������ѹ���ڻ�����ѹ��, ��Ʒ����������, ʹ��������ֲ��¶Ⱥܸߡ� ��ʱƵ��Խ��, һ�������ʱ��Խ��, ��λʱ�������Ĵ���Խ��, ����һ�λ���ʱ��������С; ��������ʱ���, ����ռ�ձȱ��ֲ����������, ÿ������������ʱ���, ����һ������Һ�и����ʱ����ȴ����, ��һ�����緢�������������١� �����������õĽ����ʹ������Ĥ��ֱ����С, ��ʴ�Եõ���ߡ� ����Ƶ��Խ��, �ں������������ѹҲԽ�ߡ� ���Ƶ�ʹ���, ��ʱ����ӳ��ᵼ�³�����Դ���Ƶĵ�ѹ��Χ, ��ʹ��Դ������[48]��
4.2 �����ܶ�
�����ܶ���Ӱ��������С����Ҫ����֮һ, ��������ŵ������ʱ�������Ĥ�ĺ�ȡ� ������ò�� ��ʴ���Լ����������ء�
���ŵ����ܶȵ�����, ����������Ӧ�����е�����������, �ṩ�����ĵ��������ʱ������, ���Ե����ܶȵ����ߵ����˲����������ʱ������[49]; ��ͬ��������ʱ����, ����Ĥ�ĺ�ȱ��ֳ�������������[4, 48-50]; �մɲ�������ڻŵ��γɵ�����������, ��������; ��ʴ�Գ��ֳ�����ǿ�͵�����[48, 49]�� �����ܶ�����, ����������Ĥ������, �ﵽͬ�����յ�ѹ����ʱ�����̡� ���ǵ����ܶȹ���, ʹ�õ�����ŵ�����̫��, ���·ŵ�ͨ����ȴ���̺����µ�������, �Ӷ�ʹ����Ĥ����ʴ���½���
Ϊ�˵õ������������������Ĥ, һЩ���ײ��÷ּ�ʽ�������Ʒ�ʽ[39, 48, 51-53], ��Shi��[51]�о��˵����ܶȲ��ζ�����Ĥ���ܵ�Ӱ��, �������Ϊ�˵õ���ʴ�Ը��õ�����Ĥ��, Ӧ���ڲ�ͬ��������ʹ�ò�ͬ�ĵ����ܶ�: �������ĵ�һ��(��ѹ0~10V)�͵ڶ���(��ѹ10~190V), ʹ�ô�ĵ����ܶ��������Ч��, ���������ĵ�����(��ѹ����190V), ʹ��С�ĵ����ܶ�ʹ����Ĥ����С�����״Ӷ��������Ĥ����ʴ�ԡ�
����һ���ڷ�Ӧǰ��ʹ�ýϴ�ĵ����ܶ�, һ�������ʹ�����������γ�����Ĥ, ��ֹ���帯ʴ, ��һ���������������ʱ�䡣 ����ڴ��������ر�����ˮ��ҵ���Զ���������˵, ������Ҫ; ���ڷ�Ӧ����ʱ, ʹ�ý�С�ĵ����ܶ�, ����ʹ������ŵ�����С, �Ӷ���������Ĥ�Ŀ�϶�������������Ʒ����ʴ�ԡ� �����ƿ�ʼʱʹ�ô�ĵ����ܶȽ�������Ȼ�͵����ܶ������ơ�
�����ܶȻ���Ӱ������Ĥ����ɡ� ��3mol/L KOH+0.6mol/L KF+0.21mol/L Na3PO4��Һ��, �������ܶ�Ϊ5mA/cm2ʱ, ����Ĥ��MgO��MgAl2O4�����������, ���������ܶ�Ϊ15mA/cm2ʱ, ����Ĥ���ɵ�һ��MgAl2O4���[14]��
����, �����ܶ�Ҳ��Ӱ������Ĥ�����ֲڶ�[50]�Լ�Ĥ����Ӳ��[54]��
4.3 �յ�ѹ
����ʹ�ú�������, Ҳ����ʽ(3)��(10)�е�im1�̶�, ����Ĥ�ĺ����Ҫ������ܶȺ�����ʱ���й�, Ҳ������Ҫ���յ�ѹ������ �����ܶ�Խ��, ����ʱ��Խ��, �յ�ѹԽ��, ����Ĥ�ĺ��Խ��
������ѹ��������Ĥ�������֯�ṹ�ͳɷ�������Ӱ��, �������Ŵ�����ѹ������, MgO�ĺ�����������[29, 30, 55]�� ������Ϊ�ڵ͵�ѹʱ, �ŵ�С, �������������¶ȵ�, ����ʩ�ӵ�ѹ�ܸ�ʱ, ���, ���������¶ȸ�, �������ơ��սᡱ������, �ߵ�������������������ˮ�Ӷ��ڻ����γ����ܵ�������[56]��
����������Ĥ�������֯�ṹ��ɷ�ֱ��Ӱ������ʴ��, ����յ�ѹ������Ĥ����ʴ���������, ������֮�䲻���ڼĶ�Ӧ��ϵ[29, 30, 55, 57-58]�� ���յ�ѹ������Ĥ��ʴ�Ե�Ӱ�����������: һ����, �յ�ѹԽ��, ����Ĥ�ĺ��Խ��; �������Ĥ�Ŀ�϶��(�ɿĴ�С�Ϳ����֮��ľ������)�������ֲ���, ������Ĥ����ʴ����Ĥ������Ӷ����; ��һ����, �յ�ѹԽ��, �����ϲ����Ļ�Խ��, ��������Ĥ��ֱ������[24], ��Ȼ��ʱ����Ĥ�ĺ��������, ���մɲ����ʴ�Է����½�[59]�� ���, �յ�ѹ������Ĥ����ʴ�Բ��ɼĶ�Ӧ��ϵ, ����������õĹ����Լ�������Ʒ��ʹ�ó���, ��������ʵ��ȷ���� ������������, ������ѹ��ʽ�������Ĥ��������, �����ʴ����[60]��
4.4 ռ�ձ�
����ռ�ձȶ�����Ĥ���ܵ�Ӱ��, Ŀǰ���о������һ�¡� �еĽ������: �ڸ�Ƶ��, ռ�ձ�Խ��, �մɲ����ֲڶ�Խ��, Խ�ֲ�; ռ�ձ�ԽС, �մɲ����ֲڶ�ԽС, Խ�⻬�� ���ڵ�Ƶ����, ռ�ձȶ��մɲ�ı���ֲڶ�Ӱ�첻����[47]�� ���ȷ��[48]���о����Ϊ��ռ�ձȵIJ�������, �մɲ�ĺ�ȱ仯����, ��ʴ�Գ����½����ơ�
��������[38]�о���ռ�ձȶ�������Ʒ��ʴ�Ե�Ӱ��, ��Ϊ���յ�ѹ���Ǻܸ�ʱ, ռ�ձ�����ʴ��֮��û��һһ��Ӧ�Ĺ�ϵ, �ؼ���������������������ƥ�䡣 ����ʵ����ʹ�ú�������, Ҳ����im1��im2�̶�, ����Ĥ�ĺ����Ҫ���յ�ѹ������ �������ܶȺ�Ƶ�ʶ��̶���, ��ռ�ձȶ�����Ĥ��ʴ�Ե�Ӱ�����������: һ����, ��ռ�ձ�Խ��, һ�����������ڹ�����ʱ���Խ��, Ҳ��������������ʱ��Խ��, ���ʹ����Ĥ��ֱ������, �Ӷ�����ʴ�Բ���; ��һ����, �����Ǻ�������, ��ƽ������i[TX-]m1�ǹ̶���, ����ʽ(7)��(11), ����ռ�ձ�Խ��ʱ, �������i1��������ʱ��ε�ƽ������ԽС, ������������Ʒ����ʴ�������� ������ �����������ۺ�������, ռ�ձ�������Ĥ����ʴ�Բ���һһ��Ӧ��ϵ��
ռ�ձȲ���Ӱ������Ĥ����ʴ��, ���������ı�����ò�йء� ���յ�ѹ�ܸ�ʱ, ռ�ձȶ�������Ʒ�����Ӱ��ܴ� ��������[38]�о�����Ƶ��600Hz�� �����ܶ�20mA/cm2�� �յ�ѹ480Vʱռ�ձȷֱ�Ϊ15%�� 25%��35%��������Ʒ�����Ӱ��, ����ռ�ձȵ�����, ����Ĥ������Խ��Խ���ɡ�
���Ͻ����˵����������Ĥ���ܵ�Ӱ��, ��Ҫǿ������, ���ڸ��о�����ʹ�õĵ��Һ��ͬ, ѡ������ʱ��������ʹ�õĵ��Һ��ƥ�䡣 ����[12]������һЩ����þ�Ͻ������������ղ�����
5 ��չ����
Mg�Ͻ�������������������������͵ķ�չ, ȡ��������ɹ�, ͬʱ��������������������������ɹ�, ������Ϊþ�Ͻ���������������ķ�չ����Ϊ:
1) ���ż����������������Դ�ϵ�Ӧ��, �����豸�ķ�չ����ΪƵ��Խ��Խ�ߺ������ܻ�, ͨ��������, ʹ������岨������, �Ա�֤����Ĥ��ͬ�����ι������ߵķֶ��趨[48, 51-52];
2) ���پ����ڴ�ͳ�ϵĺ����������ѹ����, ���������ߵ����, �������ʩ�ӽϸߵĵ�ѹ�������������һ���ĵ���, Ȼ����ڽ�������ֱ����ѹ����֤��������[24-25], Ҳ�ɲ����ڽ����ϵ���ֱ��[61], ���ɲ����������ij���ʹ�ú�����������, һ���ﵽ�趨���յ�ѹ��, �Ͳ��ú�ѹ��������[56, 62];
3) Ϊ�˵õ�������Ҫ������Ĥ, �����ۺϿ��ǹ����ķ��������Լ������֮������Լ�� ��ý�������ʵ��, �Ի����ѹ��ղ�����ϡ�
REFERENCES
[1]ʦ����, ����, ������, ��. �����ҹ�����þ��ҵ��չ�Ľ���[J]. ���ϵ���, 2001, 15(4): 5-7.
SHI Chang-xu, LI Heng-de, WANG Dian-zuo, et al. A proposal on acceleration development of metallic magnesium industry in China[J]. Materials Review, 2001, 15(4): 5-7.
[2]Froes F H, Eliezer D, Aghion E. The science, technology, and applications of magnesium[J]. JOM, 1998, 5(9): 30-34.
[3]Decker R F. The renaissance in magnesium[J]. Advanced Materials and Processes, 1998, 154(3): 31-33.
[4]Sharma A K, Rani R U, Giri K. Studies on anodization of magnesium alloy for thermal control applications[J]. Metal Finishing, 1997, 95(3): 43-51.
[5]Gray J E, Luan B. Protective coatings on magnesium and its alloys��A critical review[J]. Journal of Alloys and Compounds, 2002, 336: 88-113.
[6]���ٷ�, ������, ������, ��. þ�Ͻ����������о���չ��չ��[J]. �й���ɫ����ѧ��, 2006, 16(7): 1136-1148.
ZHANG Rong-fa, SHAN Da-yong, HAN En-hou, et al. Status and prospect of anodization on magnesium and its alloys[J]. The Chinese Journal of Nonferrous Metals, 2006, 16(7): 1136-1148.
[7]������, �ϴ�ΰ, ������, ��. þ��þ�Ͻ��������������Һ���乤��[J]. ���ϱ���, 2002, 35(3): 39-41.
ZHANG Yong-jun, YAN Chuan-wei, WANG Fu-hui, et al. Environmental-friendly bath solution and process for anodization of magnesium and its alloys[J]. Materials Protection, 2002, 35(3): 39-41.
[8]����ѫ. ʵ�õ�Ƽ���[M]. ����: ��ѧ��ҵ������, 2002: 17.
HUANG Zi-xun. Applied Plating Technology[M]. Beijing: Chemical Industry Press, 2002: 17.
[9]Verdier S, Boinet M, Maximovitch S, et al. Formation, structure and compositions of anodic films on AM60 magnesium alloy obtained by DC plasma anodizing[J]. Corrosion Science, 2005, 47: 1429-1444.
[10]Yerokhin A L, Nie X, Leyland A, et al. Plasma electrolysis for surface engineering[J]. Surface and Coatings Technology, 1999, 122: 73-93.
[11]Beauvir J. Oxidizing Electrolytic Method for Obtaining a Ceramic Coating at the Surface of a Metal[P]. US Patent: 6808613, 2004-10-26.
[12]Kuhn A. Plasma anodizing of magnesium alloys[J]. Metal Finishing, 2003, 101(9): 44-50.
[13]Khaselev O, Yahalom J. The anodic behavior of binary Mg-Al alloys in KOH-aluminate solutions [J]. Corrosion Science, 1998, 40(7): 1149-1160.
[14]Khaselev O, Weiss D, Yahalom J. Anodizing of pure magnesium in KOH-aluminate solutions under sparking [J]. J Electrochem Soc, 1999, 146(5): 1757-1761.
[15]Khaselev O, Weiss D, Yahalom J. Structure and composition of anodic films formed on binary Mg-Al alloys in KOH-aluminate solutions under continuous sparking [J]. Corrosion Science, 2001, 43: 1295-1307.
[16]Bonilla F A, Berkani A, Skeldon P, et al. Enrichment of alloying elements in anodized magnesium alloys[J]. Corrosion Science, 2002, 44: 1941-1948.
[17]Bonilla F A, Berkani A, Liu Y, et al. Formation of anodic films on magnesium alloys in an alkaline phosphate electrolyte[J]. J Electrochem Soc, 2002, 149(1): B4-B13.
[18]Ono S, Asami K, Osaka T, et al. Structure of anodic films formed on magnesium[J]. J Electrochem Soc, 1996, 143(3): L62-L63.
[19]Ѧ�ı�, ��־��, ������, ��. ZM5þ�Ͻ�������Ĥ����������[J]. �����ȴ���ѧ��, 1998, 19(3): 42-45.
XUE Wen-bin, DENG Zhi-wei, LAI Yong-chun, et al. Growth regularity of ceramic film in microarc oxidation on cast magnesium alloy[J]. Transactions of Metal Heat Treatment, 1998, 19(3): 42-45.
[20]Ѧ�ı�, ��־��, ��ͨ��, ��. ����þ�Ͻ�����������[J]. ϡ�н��������빤��, 1999, 28(6): 353-356.
XUE Wen-bin, DENG Zhi-wei, ZHANG Tong-he, et al. Microarc oxidation mechanism of a cast magnesium alloy[J]. Rare Metal Materials and Engineering, 1999, 28(6): 353-356.
[21]���, ���Ҳ�, ������, ��. ��ͬ�����Һ�������������е�����[J]. �й���ʴ�����ѧ��, 2004, 24(4): 222-225.
LI Jian-zhong, SHAO Zhong-cai, TIAN Yan-wen, et al. The action of the forms of P element on the process of the micoarc oxidation[J]. Journal of Chinese Society for Corrosion and Protection, 2004, 24(4): 222-225.
[22]Zozulin A J, Bartak D E. Anodized coatings for magnesium alloys[J]. Metal Finishing, 1994, 92(3): 39-44.
[23]McNeill W. The Cr-22 coating for magnesium[J]. Metal Finishing, 1955, 53(12): 57-59.
[24]Birss V, Xia S, Yue R, et al. Characterization of oxide films formed on Mg-based WE43 alloy using AC/DC anodization in silicate solutions[J]. J Electrochem Soc, 2004, 151(1): B1-B10.
[25]Xia S J, Yue R, Rateick R G Jr, et al. Electrochemical studies of AC/DC anodized Mg alloy in NaCl solution[J]. J Electrochem Soc, 2004, 151(3): B179-B187.
[26]Mato S, Alcala G, Skeldon P, et al. High resistivity magnesium-rich layers and current instability in anodizing a Mg/Ta alloy[J]. Corrosion Science, 2003, 45: 1779-1792.
[27]Abulsain M, Berkani A, Bonilla F A, et al. Anodic oxidation of Mg-Cu and Mg-Zn alloys[J]. Electrochimica Acta, 2004, 49: 899-904.
[28]Khaselev O, Yahalom J. Constant voltage anodizing of Mg-Al alloys in KOH-Al(OH)3 solutions[J]. J Electrochem Soc, 1998, 145(1): 190-193.
[29]Mizutani Y, Kim S J, Ichino R, et al. Anodizing of Mg alloys in alkaline solutions[J]. Surface and Coatings Technology, 2003, 169-170: 143-146.
[30]Kim S J, Okido M, Mizutani Y, et al. Formation of anodic films on Mg-Al alloys in NaOH solutions at constant potentials[J]. Materials Transactions, 2003, 44(5): 1036-1041.
[31]Fukuda H, Matsumoto Y. Effects of Na2SiO3 on anodization of Mg-Al-Zn alloy in 3mol/L KOH solution[J]. Corrosion Science, 2004, 46: 2135-2142.
[32]Grubbs C A. Anodizing by current density-an update[J]. Metal Finishing, 1999, 97(9): 71-73.
[33]������. þ��þ�Ͻ�����������������Լ����о�[D]. ����: �й���ѧԺ�����о���, 2003: 49-65.
ZHANG Yong-jun. Study on Environmentally Friendly Anodizing Surface Modification for Magnesium and Its Alloys[D]. Shenyang: Institute of Metal Research, Chinese Academy of Sciences, 2003: 49-65.
[34]Schmeling, Edith L, Roschenbleck, et al. Method of Preparing the Surfaces of Magnesium and Magnesium Alloys[P]. US Patent: 4976830, 1990-12-11.
[35]Schmeling, Edith L, Roschenbleck, et al. Method of Producing Protective Coatings that are Resistant to Corrosion and Wear on Magnesium and Magnesium Alloys[P]. US Patent: 4978432, 1990-12-18.
[36]Bartak, Duane E, Lemieux, et al. Hard Anodic Coating for Magnesium Alloys[P]. US Patent: 5470664, 1995-11-28.
[37]Kurze, Peter, Banerjee, et al. Method of Producing Oxide Ceramic Layers on Barrier Layer-Forming Metals and Articles Produced by the Method[P]. US Patent: 5385662, 1995-01-31.
[38]���ٷ�. þ�Ͻ������������ż��ʴ���о�[D]. ����: �й���ѧԺ�����о���, 2005: 52-79.
ZHANG Rong-fa. Study of Anodization and Galvanic Corrosion in Magnesium Alloys[D]. Shenyang: Institute of Metal Research, Chinese Academy of Sciences, 2005: 52-79.
[39]WANG Li-shi, CAI Qi-zhou, WEI Bo-kang, et al. Characterization of oxide films formed on magnesium alloys using bipolar pulse microarc oxidation in phosphate solutions[J]. Trans Nonferrous Met Soc China, 2005, 15(3): 600-605.
[40]BurgerF J, Wu J C. Dielectric breakdown in electrolytic capacitors[J]. J Electrochem Soc, 1971, 118(12): 2039-2042.
[41]Vijh A K. Sparking voltages and side reactions during anodization of valve metals in terms of electron tunneling[J]. Corrosion Science, 1971, 11: 411-417.
[42]Wood G C, Pearson C. Short communication of dielectric breakdown of anodic oxide films on valve metal[J]. Corrosion Science, 1967, 7: 119-125.
[43]Yahalom J, Hoar T P. Galvanostatic anodizing of aluminum[J]. Electrochimica Acta, 1970, 15: 877-884.
[44]Ikonopisov S. Theory of electrical breakdown during formation of barrier anodic films[J]. Electrochimica Acta, 1977, 22: 1077-1082.
[45]Yahalom J, Zahavi J. Experimental evaluation of some electrolytic breakdown hypotheses[J]. Electrochimica Acta, 1971, 16: 603-607.
[46]�Ž�, ���ں�, ���ٲ�, ��. þ�Ͻ�Ӧ��[M]. ����: ��ѧ��ҵ������, 2004: 231.
ZHANG Jin, ZHANG Zong-he, ZENG Rong-chang, et al. Magnesium Alloys and Applications[M]. Beijing: Chemical Industry Press, 2004: 231.
[47]�½���, �º�, ���پ�. �������þ�Ͻ��������մɲ������Ժ͵绯ѧ�迹��Ӱ��[J]. ��ʴ�����, 2003, 24(6): 249-251.
HAO Jian-min, CHEN Hong, ZHANG Rong-jun. Effects of electric parameters on density and electrochemical impedance of ceramic layer made by micro-arc oxidazation on magnesium alloy[J]. Corrosion & Protection, 2003, 24(6): 249-251.
[48]���ȷ�, ������. ����������þ�Ͻ��������մɲ���ʴ�Ե�Ӱ��[J]. ��ʴ��ѧ���������, 2005, 17(3): 141-143.
ZHANG Xian-feng, JIANG Bai-ling. Effects of energy parameters on corrosion resistance of micro-arc oxidation coatings on magnesium alloys[J]. Corrosion Science and Protection Technology, 2005, 17(3): 141-143.
[49]�����, ��ï��, ��ݷ, ��. �����ܶȶ�þ�Ͻ����������̼������մ�Ĥ���ܵ�Ӱ��[J]. ϡ�н��������빤��, 2005, 34(10): 1554-1557.
GUO Hong-fei, AN Mao-zhong, XU Shen, et al. Effects of current density on mechanism of micro-arc oxidation and property of ceramic coating formed on magnesium alloys[J]. Rare Metal Materials and Engineering, 2005, 34(10): 1554-1557.
[50]����, ������,���, ��. ���ղ�����AZ91Cþ�Ͻ�����������Ĥ���Ӱ��[J]. ���Ͽ�ѧ�빤��ѧ��, 2005, 23(2): 262-265.
LIAN Jun, GUO Bao-gang, TIAN Jun, et al. Influence of process parameters on microarc oxidation coatings on AZ91C magnesium alloys[J]. Journal of Materials Science & Engineering, 2005, 23(2): 262-265.
[51]Shi Z, Song G, Atrens A. Influence of anodising current on the corrosion resistance of anodised AZ91D magnesium alloy[J]. Corrosion Science, 2006, 48(8): 1939-1959.
[52]������, ���ȷ�. þ�Ͻ��������մɲ���������̼�����ʴ��[J]. �й���ʴ�����ѧ��, 2005, 25(2): 97-101.
JIANG Bai-ling, ZHANG Xian-feng. Growth process and corrosion resistance of ceramic coatings formed by micro-arc oxidation on magnesium alloy[J]. Journal of Chinese Society for Corrosion and Protection, 2005, 25(2): 97-101.
[53]Cai Q, Wang L, Wei B, et al. Electrochemical performance of miroarc oxidation films formed on AZ91D magnesium alloy in silicate and phosphate electrolytes[J]. Surface and Coatings Technology, 2006, 200(12-13): 3727-3733.
[54]��ʤ��, ����, �ܺ���, ��. þ�Ͻ����������������о�[J]. ���ϴ�ѧѧ��(��Ȼ��ѧ��), 2005, 32(3): 15-18.
LUO Sheng-lian, DAI Lei, ZHOU Hai-hui, et al. Research on environmental-friendly anodizing process for magnesium alloys[J]. Journal of Hunan University (Natural Sciences), 2005, 32(3): 15-18.
[55]Ҧ����, �ܰ���, ������. ��ѹ��þ�Ͻ�������Ĥ��֯����ʴ�Ե�Ӱ��[J]. ���ϱ���, 2005, 38(6): 7-10.
YAO Mei-yi, ZHOU Bang-xin, WANG Jun-an. Effect of working voltage on microstructure and corrosion resistance of micro-arc oxidation coating on MB5 magnesium alloy[J]. Materials Protection, 2005, 38(6): 7-10.
[56]Hsiao H Y, Tsai W T. Characterization of anodic films formed on AZ91D magnesium alloy[J]. Surface and Coatings Technology, 2005, 190: 299-308.
[57]Blawert C, Heitmann V, Dietzel W, et al. Influence of process parameters on the corrosion properties of electrolytic conversion plasma coated magnesium alloys[J]. Surface and Coatings Technology, 2005, 200: 68-72.
[58]���, ����, �żʱ�. þ�Ͻ������������в�ͬ��ѹ�»��Ĥ��������о�[J]. �й���ʴ�����ѧ��, 2005, 25(5): 267-270.
WANG Yan-hua, WANG Jia, ZHANG Ji-biao. Properties of anodic coatings on AZ91D Mg alloys during micro-arc oxidation process[J]. Journal of Chinese Society for Corrosion and Protection, 2005, 25(5): 267-270.
[59]�½���, �º�, ���پ�, ��. þ�Ͻ��������մɲ����ʴ��[J]. �й���ɫ����ѧ��, 2003, 13(4), 988-991.
HAO Jian-min, CHEN Hong, ZHANG Rong-jun, et al. Corrosion resistance of magnesium alloy micro-arc oxidization ceramic coating[J]. The Chinese Journal of Nonferrous Metals, 2003, 13(4): 988-991.
[60]��ΡΡ, ����, ������, ��. þ�Ͻ�����������������̿��Ƶ��о�[J]. �����ȴ���ѧ��, 2005, 26(1): 77-80.
GONG Wei-wei, ZHANG Le, WU Xiao-ling, et al. Controlling process of plasma microarc oxidation on magnesium alloy[J]. Transactions of Materials and Heat Treatment, 2005, 26(1): 77-80.
[61]Timoshenko A V, Magurova Y V. Investigation of plasma electrolytic oxidation processes of magnesium alloy MA2-1 under pulse polarisation modes[J]. Surface and Coatings Technology, 2005, 199: 135-140.
[62]Hsiao H Y, Tsung H C, Tsai W T. Anodization of AZ91D magnesium alloy in silicate-containing electrolytes[J]. Surface and Coatings Technology, 2005, 199: 127-134.
�ո�����: 2006-04-26; ������: 2006-08-15
ͨѶ����: ���ٷ�, ����; �绰: 0791-3801423; E-mail: rfzhang-10@163.com

