文章编号:1004-0609(2009)09-1625-07
稀土铝合金Miedema模型计算中的参数φ*
占春耀,王 为,汤振雷,聂祚仁
(北京工业大学 材料科学与工程学院,北京 100022)
摘 要:研究修改计算式中电负性参数φ*,考虑合金化过程中不同元胞边界上电子化学势差会对合金形成能产生负的贡献,提出Miedema模型中参数φ*的计算表达式。结果表明:参数φ*的计算值与功函数值比较接近,与功函数值相比,其最大相对误差在12.0%以内,平均误差低于5.0%,计算得到的稀土铝合金系统混合焓与实验值更接近。
关键词:铝合金;稀土元素;Miedema模型;功函数;混合焓
中图分类号:TG 111.5 文献标识码:A
Miedema model calculation of parameter φ* for aluminum alloys with rare earth elements
ZHAN Chun-yao, WANG Wei, TANG Zhen-lei, NIE Zuo-ren
(School of Materials Science and Engineering, Beijing University of Technology, Beijing 100022, China)
Abstract: The difference of the electron chemical potential energy between the atom cells in the calculated aluminum alloys was considered to have a negative effect on the formation energy of the alloy system. The function of parameter φ* to the thermodynamic calculation was assessed and a new formula about the parameter φ* for rare earth elements were established. The results show that the calculated value through the proposal parameter φ* is near to the system work function, and the maximum relative error is within 12.0% while the average relative error is less than 5.0%. The calculated mixing enthalpy values of the alloys are much closer to the published experimental results.
Key words: aluminum alloys; rare earth elements; Miedema model; work function; mixing enthalpy
合金热力学性质的研究对合金生产、应用及基础理论研究具有重要意义。由于高温实验条件的限制,使得通过实验条件来研究某些合金的相变过程变得困难,而且也不能确保研究结果的准确性。因此,利用数学模型方法计算就成为热力学研究的重要手段之 一[1]。自1950年以来,材料学者们建立了许多理论模型,其中最成功的一种实用理论是采用简单物理参数所形成的Miedema模型,该模型采用摩尔体积V、Wiger-SeitZ原子胞边界电子密度nws以及电负性参数φ* 3个物理参数。多年来应用该模型的实际效果较令人满意[2-5],但是,CHEN等[6-7]在研究金属间化合物的Laves相时发现,Miedema模型的计算结果不适用于锆基合金。对此深入研究后,发现其误差主要来自于Miedema模型中参数φ*取值与其功函数值相差较大。元素的功函数是指在热电辐射或光电辐射中,从金属表面移走一个电子所需的能量,通过对锆元素φ*参数的修正,取得了满意的结果[8]。余胜文等[9]对稀土铝合金的热力学性能和高温性能的研究中采用热力学计算的方法时也存在较大的偏差。作为对铝合金中合金元素作用机理的进一步综合考虑,本文作者提出关于稀土元素的Miedema模型参数φ*的计算表达式。采用CHEN等[8]修正的Miedema模型参数φ*作为对比计算,并结合铝合金的相变机理和结构特点,讨论不同条件得出的参数φ*值对铝稀土合金系统混合焓产生的影响,给出适用于稀土元素的Miedema计算模型中参数φ*的计算表达式。
1 热力学计算模型和参数
1.1 Miedema计算模型中参数φ*的取值分析
在Miedema模型中用来表征元素最基本物理本质的3个参数分别为摩尔体积V、Wiger-SeitZ原子胞边界电子密度nws和电子化学势参数φ*,其中元素摩尔体积V的数据基本上得到了实验的验证,并有标准数据可查。电子密度nws与(B/V)1/2有关,也已经很好地予以定义。因此,电子密度和摩尔体积这两个参数基本可以确定。然而,第三个参数的定义与取值一直存在着争议。在原来的Miedema模型中参数φ*取值一般认为由实验测定的功函数的平均值确定,早期MIEDEMA等[10]认为参数φ*与元素的功函数值之间存在着一定的联系,所以有时也称参数φ*为功函数标。在之后对多体系合金的计算研究中逐步增加经验,到1983年针对模型计算的形成热与实验值的情况[11-15],对φ*值进行过调整。由于其考虑的物理机制各有不同,一方面MIEDEMA认为φ*与电负性Xp具有很好的线性关系[16],因此有时φ*被称为电负性参数,这是大部分文献中都采用的说法;另一方面,MIEDEMA等又根据HODGES和SCOTT[17]提出的金属元素的电子化学势μ与电负性Xp具有近似线性关系的事实,认为元素的功函数与电子化学势之间具有相当好的线性关系,因此也可以称φ*为电子化学势参数。
由此可知,Miedema模型参数φ*的确定没有完全的物理机理的认识,还是带有经验性的。CHELIKOWSKY和PHILLIPS[18]基于量子亏损理论所得到的与轨道角动量有关的原子实半径ri值,从理论上导出了27种简单金属的Miedema模型参数φ*的表达式:
 但是在实际的应用中,有些合金元素的数据难以得到,一些稀土元素的则更难计算。到目前为止,国内外的学者基本上是采用MIEDEMA等提供的参数φ*值。综合来看,Miedema模型中φ*参数与元素的功函数值相关。CHEN等[8]将Zr元素的参数φ*值由原来的3.45修正到3.62,取值更加接近功函数值,因此,其计算结果与实验值更接近。
但是在实际的应用中,有些合金元素的数据难以得到,一些稀土元素的则更难计算。到目前为止,国内外的学者基本上是采用MIEDEMA等提供的参数φ*值。综合来看,Miedema模型中φ*参数与元素的功函数值相关。CHEN等[8]将Zr元素的参数φ*值由原来的3.45修正到3.62,取值更加接近功函数值,因此,其计算结果与实验值更接近。
1.2 参数φ*表达式的提出
稀土元素比较特殊,除尺寸因素之外,还具有特殊的原子和离子状态的电子组态,在含稀土铝合金的热力学计算中,采用Miedema模型时的φ*参数选择有重大的影响,因此,对正确预测含稀土铝合金的热力学性能产生较大的偏差。对比利用本文作者提出的计算式得出的和原始的参数φ*值计算出的稀土铝合金系统的混合焓值可知,两种不同的参数φ*取值会使Miedema模型的计算结果有很大的偏差,其中Eu、Yb元素分别与Al元素形成的合金系统的混合焓值相差最大达到58和28 kJ/mol。
一般认为影响稀土元素铝合金热力学计算偏差的主要因素是尺寸和元素的原子及离子组态的差别。从化学价键原理来看,稀土元素的4f轨道并未填满,Al元素的3p轨道也未填满,3p轨道的能量又低于稀土元素的5s轨道的能量,这样Al原子提供轨道,稀土原子提供电子,轨道会发生简并,使得形成的合金体系的结构发生变化,因而必须考虑在使用Miedema模型计算稀土元素和铝形成合金系统时其原子和离子组态对参数φ*的影响。本文作者在综合考虑稀土元素特殊的原子和离子组态的基础上,借鉴文献[8]对参数φ*的表达形式提出适用于稀土元素的参数φ*的表达式:

一般镧系离子常价态为+3价,其中Eu、Yb取+2价;对于参数a和b的确定,首先是假定Sc、Y元素的参数φ*与其功函数值一致,然后通过线性回归处理得到参数a和b分别为0.157 5和2.014 4,得到式(3):
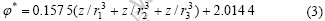
表1 不同配位体时稀土元素的离子半径[19-20]
Table 1 Ion radius of rare earth elements with different coordination numbers[19-20] (pm)
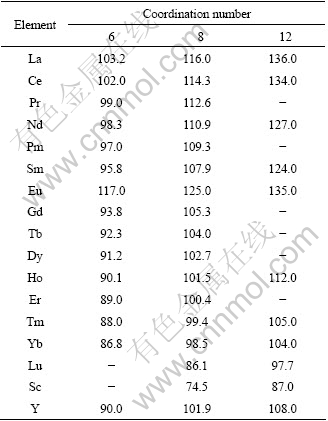
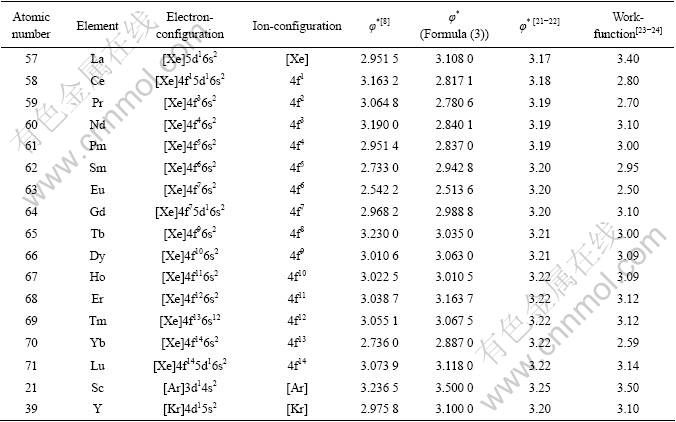
因为镧系稀土元素的性质相似,因而可以利用式(3)来计算其他稀土元素的参数φ*值,结果如表2所列。
表2 稀土元素的电子构型与计算参数φ*和功函数值之间的关系
Table 2 Relationship among electron and ion configuration of rare elements, Miedema model parameter φ* and work functions
1.3 Miedema生成热计算模型
关于热力学性能的计算,本文作者采用NIESSEN等[21]提出的Miedema模型计算式,其表达式如下:

 依余胜文等[9]采用的稀土铝合金体系相关参数的取值原则,将计算过程编程计算出稀土铝二元合金固态或液态合金的生成热。
依余胜文等[9]采用的稀土铝合金体系相关参数的取值原则,将计算过程编程计算出稀土铝二元合金固态或液态合金的生成热。
2 结果与讨论
2.1 参数φ*值
由表2可以明显看出,文献[21-22]中采用的稀土元素参数φ*值比较接近,几个元素的取值基本相同,如Pr、Nd、Pm的参数φ*都为3.19。数据的取值虽然和稀土元素的性质比较相似,但是这些元素的电子组态和离子组态存在着一定的本质性差别,因而对计算结果产生一定的影响,对于正确的预测和评价合金体系的热力学性质产生一定的偏差[9, 14-15]。本文作者基于以上考虑提出了适用于稀土元素的参数φ*的计算表达式。图1所示为3种不同的针对稀土元素的参数φ*的计算值与功函数值相比得到的相对误差结果。从图1中可以明显看到,与利用文献[8]中提供的参数φ*表达式计算出的值以及文献[21-22]提供的数据相比,通过式(3)计算出的参数φ*值更接近实验功函数值,相对误差最大不超过12.0%,平均误差低于5.0%。
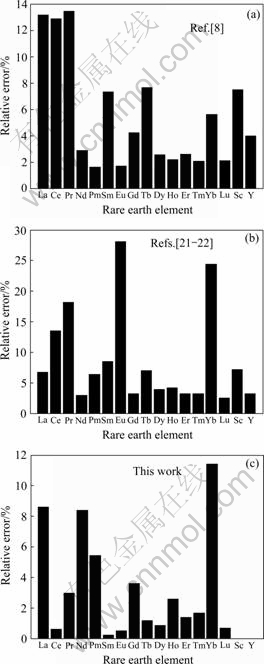
图1 3种不同方法得到的稀土元素的参数φ*值与功函数值相比得到的相对误差结果
Fig.1 Relative errors between work function and parameter φ* value from Ref.[8](a), Refs.[21-22](b) and this work(c)
2.2 混合焓值
图2~8所示为铝和稀土元素形成的合金系统的混合焓随稀土元素含量变化的规律,其中包括铝与17种稀土元素形成的合金系统的混合焓的计算结果。与文献[21-22]提供的参数φ*值计算的结果相比,由本文修正的参数φ*值计算的结果与实验值符合得较好。从表2中可以看出,修正前后元素Yb、Eu的参数φ*值相差分别为0.38和0.66,与其功函数之值相差分别为0.63和0.70。从图7~8可以看出,修正前后计算出来的合金系统的混合焓相差分别达到了58和28 kJ/mol,这也说明Miedema模型的参数φ*是一个对结构指数敏感性强的参数。

图2 1 200 K时Al-La合金的混合焓随La含量的变化曲线
Fig.2 Variation of mixing enthalpy with La content in Al-La alloys at 1 200 K

图3 1 200 K时Al-Ce合金的混合焓随Ce含量的变化曲线
Fig.3 Variation of mixing enthalpy with Ce content in Al-Ce alloys at 1 200 K

图4 1 873 K时 Al-Sc合金的混合焓随Sc含量的变化曲线
Fig.4 Variation of mixing enthalpy with Sc content in Al-Sc alloys at 1 873 K

图5 1 550 K时 Al-Nd合金的混合焓随Nd含量的变化曲线
Fig.5 Variation of mixing enthalpy with Nd content in Al-Nd alloys at 1 550 K
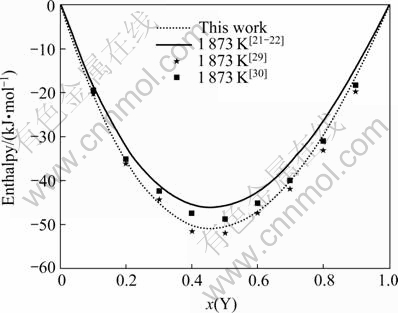
图6 1 873 K时Al-Y合金的混合焓随Y含量的变化曲线
Fig.6 Variation of mixing enthalpy with Y content in Al-Y alloys at 1 873 K
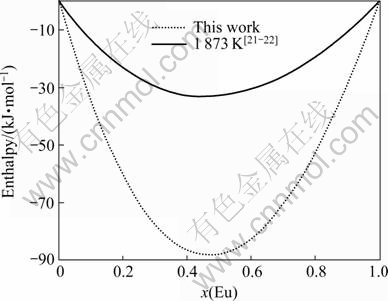
图7 1 873 K时Al-Eu合金的混合焓随Eu含量的变化曲线
Fig.7 Variation of mixing enthalpy with Eu content in Al-Eu alloys at 1 873 K
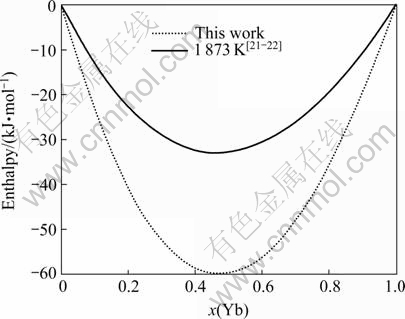
图8 1 873 K时Al-Yb合金的混合焓随Yb含量的变化曲线
Fig.8 Variation of mixing enthalpy with Yb content in Al-Yb alloys at 1 873 K
对比图2~8可以看出,Miedema参数φ*值的改变对合金系统混合焓会产生较大的影响,尤其是Al-Yb、Al-Eu合金系统。ZHU等[1]认为可能是由稀土元素本身特殊的电子结构所引起,它们的化合价为+2价,在和铝之间的交互作用过程中降低了总的键能,因而降低了其与铝形成的合金系统中的混合焓。Miedema模型中参数φ*与元素的功函数密切相关[10, 17],推论认为影响稀土元素功函数的因素也会间接影响参数φ*的取值。
根据能量最低原理,镧系元素自由原子的基态电子组态有两种类型:[Xe]4fn6s2和[Xe]4fn-15d16s2,各稀土元素原子的基组态如表2所列。图9所示为镧系元素原子组态相对能量大小分布图。从图9和表2可以看出,随着f层电子的增加,原子组态能量相对值总的趋势是增加的,当接近半充满或全充满时,原子组态能量相对值突然增加,功函数的变化规律正好相反。稀土原子最内层是[Xe]结构的饱和层,最外层是两个电子,4f层是未饱和的电子轨道层,从镧原子到镥原子4f层电子个数依次增加,即从轻稀土到重稀土4f电子有所不同。由于内层电子在成键中也会起到作用,产生p-f杂化(轨道简并),电子结构和数量不同,杂化效果也会不同。从化学价键原理来看,稀土元素化学性质很活泼,而Al元素的3p轨道并未填满,3p轨道的能量又低于稀土元素的5s轨道的能量,这样Al原子提供轨道,稀土原子提供电子,能够相互结合,二者相互作用时轨道会发生简并,因而稀土元素与铝合金化的过程中各自的电子和离子组态都要发生变化,这一点自然体现在与原子和离子组态相关的参数φ*上,其表现是越接近功函数值,计算出的铝稀土系统混合焓值越接近实验值。相反,从图9可以看出,Eu、Yb原子组态的相对能量最高,电子挣脱原子核作用所需要的能量与原子组态的相对能量成反比,因而Eu、Yb元素的功函数相对较低,这一点也可以从表2的数据得以证实。由于Yb、Eu原子和离子组态的特殊性,在和铝结合成合金时的系统混合焓对参数φ*的取值也越敏感,因而参数φ*的改变对系统的混合焓的影响很大(见图7和8)。鉴于稀土元素复杂的电子结构,与铝形成合金时的状态更加复杂,因而在参数φ*的取值上要全面考虑。本文在综合考虑稀土元素的电子结构的基础上提出了Miedema模型参数φ*的新计算表达式,系列计算的稀土铝合金系统的混合焓值表明与实验值基本接近,这也说明本计算方法的合理性和可行性。
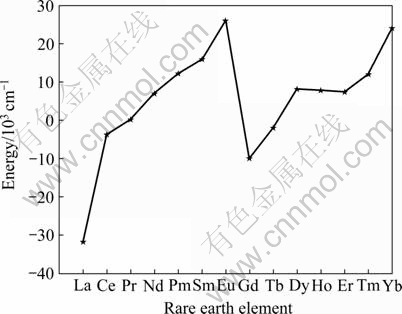
图9 镧系元素原子组态相对能量大小分布曲线[32]
Fig.9 Relative energy distribution diagram of lanthanide atoms’ configuration[32]
3 结论
1) 通过稀土元素结构特点的讨论和参数计算方法的分析比较,提出与稀土元素电子结构相关的Miedema模型参数φ*的新的计算式φ*=0.157 5×[z/r13+ z/r23+ z/r33]+2.014 4。对比文献资料表明,计算的结果与实测的功函数值基本接近,计算得到的稀土铝合金系统的混合焓值与实验值也较接近,由此证明所提出参数计算式准确性高。
2) 计算稀土铝合金的Miedema 模型中参数φ*值的微小变化,对合金系统混合焓值的影响很大,表明参数φ*是一个对合金组成和结构敏感的参数。
REFERENCES
[1] ZHU J H, LIU T, PIKE L M, LIAW P K. Enthalpies of formation of binary Laves phases[J]. Intermetallics, 2002, 10(6): 579-595.
[2] LIU B X, LAI W S, ZHANG Q. Irradiation induced amorphization in metallic multilayers and calculation of glass- forming ability from atomistic potential in the binary metal systems[J]. Mater Sci Eng R: Reports, 2000, 29(1/2): 1-48.
[3] WANG J W, GUO Q T, KLEPPA O J. Standard enthalpies of formation of some Th alloys with Group Ⅷ elements (Co, Ni, Ru, Rh, Pd, Ir and Pt) determined by high-temperature direct synthesis calorimetry[J]. J Alloy Compd, 2000, 313(1/2): 77-84.
[4] GUO Q T, KLEPPA O J. The standard enthalpies of formation of the compounds of early transition metals with late transition metals and with noble metals as determined by Kleppa and co-workers at the University of Chicago―A review[J]. J Alloy Compound, 2001, 321(2): 169-182.
[5] BOER F R, BOOM R, MATTENS W C, MIEDEMA A R, NIESSEN A K. Cohesion in metals[J]. Transition Metal Alloys, 1988, 1: 758-767.
[6] CHEN X Q, WOLF W, PODLOUCKY R, ROGL P. Ab initio study of ground-state properties of the Laves phase compounds TiCr2, ZrCr2, and HfCr2[J]. Phys Rev, 2005, B71: 174101-174108.
[7] CHEN X Q, WOLF W, PODLOUCKY R, ROGL P. Ab initio study of ground-state properties of the Laves-phase compound ZrMn2[J]. Phys Rev B, 2005, 72(5): 054440-054448.
[8] CHEN X Q, PODLOUCKY R. Miedema’s model revisted[J]. Computer Coupling of Phase Diagrams and Thermo Chemistry, 2006, 30(3): 266-269.
[9] 余胜文, 王 为. Al-Mg-Sc合金中热力学平衡相的计算[J]. 中国有色金属学报, 2006, 16(3): 505-510.
YU Sheng-wen, WANG Wei. Equilibrium phases and phase transformation of Al-Mg-Sc alloy through thermodynamic calculation[J]. The Chinese Journal of Nonferrous Metals, 2006, 16(3): 505-510.
[10] MICHAELSON H B. Relation between an atomic electronegativity scale and work function[J]. IBM J Res Develop, 1978, 22: 72-80.
[11] MIEDEMA A R, DEBOER F E, DECHATEL PF. Empirical description of the role of electronegativity in alloy formation[J]. J Phys F: Met Phys, 1973, 3: 1558-1566.
[12] MIEDEMA A R. Energy effects and charge transfer in metal physics modeling in real space[J]. Physica B: Condensed Matter, 1992, 182(1): 1-17.
[13] BOOM R, DEBOER FR, MIEDEMA A. Heat of mixing of liquid alloys[J]. J Less Commom Met, 1976, 45: 237-240.
[14] MIEDEMA A R, DE CHATEL P F. Thoery of alloy phase formation[J]. The Met Soc of AIME, 1980, 19: 344-349.
[15] MIEDEMA A R, BOOM R, DEBOER F R. Predicting heat effects in alloys[J]. Physica, 1981, 103B: 67-75.
[16] PAULING L. The nature of the chemical bond[M]. Ithaca: Cornell Univ Press, 1960.
[17] HODGES C H, SCOTT M J. Theory of electrochemical effects in alloys[J]. Phil Mag, 1972, 26: 375-380.
[18] CHELIKOWSKY J R, PHIILLIPS J C. Quantum-defect theory of heats of formation and structural transition energies of liquid and solid simple metal-alloys and compounds[J]. Phys Rev B, 1978, 17: 2453-2477.
[19] SHANNON R D, PREWITT C T. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides[J]. Acta Crystallographyca B, 1969, 25: 925-946.
[20] SHANNON R D. Revised effective ionic radii in oxides and fluorides[J]. Acta Crystallographyca A, 1976, 32: 751-767.
[21] NIESSEN A K, BOER F R, BOON B. Model prediction for the enthalpy of formation of transition metal alloy (Ⅱ)[J]. Calphad: Computer Coupling of Phase Diagrams and Thermo Chemistry, 1983, 7(1): 51-70.
[22] MIEDEMA A R, BOOM R. On the heat of formation of solid alloys[J]. Journal of the Less Common Metals, 1975, 41(2): 283-298.
[23] TRASATTI S. Individual solvated properties and specificity of ion adsorption[J]. J Chem Soc Faraday Trans Ⅰ, 1972, 68: 229-234.
[24] LANG N D, KOHN W. Theory of Metal surfaces: Work function[J]. Phys Rev, 1971, B3: 1215-1223.
[25] ESIN YU O, KOLESNIKOV S P, BAEV V M, PETRUSHEVSKII M S, GELD P V. The enthalpies of formation of liquid binary aluminium-lanthanum and tin-lanthanum alloys[J]. J Phys Chem, 1981, 55: 893-894.
[26] SOMMER F, KEITA K, KRULL H G, PREDEL B. Thermodynamic investigation of Al-La alloys[J]. J Less Common Met, 1988, 137(1/2): 267-275.
[27] ZVIADADZE G N, CHKHIKVADZE L, KERESELIDZE M V. Thermodynamic properties of aluminium-rare earth element binary melts[J]. J Phys Chem, 1976, 81(11): 149-152.
[28] ENSIN YU O, RYSS G M, GELD P V. Enthalpies of formation of cerium-aluminium molten alloys[J]. Termodin Met Splavov (Rasplavy), 1979, 2: 53-56.
[29] RYSS G M, ESIN YU O, STROGANOV A I, GELD P V. Enthalpy of formation of yttrium-aluminium molten alloys[J]. J Phys Chem, 1976, 50(44): 985-986.
[30] ESIN YU O, GELD P V, PETRUSHESKII M S, BOBROV N P, RYSS G H, SOKOLOV VV, PAZDNIKOV I P, DUBROVSKII A YA, SPLAVY V SB, SPLAVY REDK. Thermodynamic calculations in the Y-Al alloys system[J]. Journal of Alloys and Compounds, 1975: 177-182.
[31] LITOVSKII V V, VALISHEV M G, ESIN Y U O, GELD P V, PETRUSHEVSKII M S. Zh. Enthalpy of formation of liquid binary alloys of aluminium with scandium[J]. J Phys Chem, 1986, 60(9): 2310-2311.
[32] 张若桦. 稀土元素化学[M]. 天津: 天津科学技术出版社, 1987: 2-17.
ZHANG Ruo-hua. Chemistry of rare earth elements[M]. Tanjin: Tianjin Science Technology Press, 1987: 2-17.
基金项目:国家重点基础研究发展计划资助项目(2005 CB 623706);国家杰出青年科学基金资助项目(50525413)
收稿日期:2008-01-04;修订日期:2008-05-15
通信作者:聂祚仁,教授,博士;电话:010-67391536;E-mail: zrnie@bjut.edu.cn
(编辑 李向群)

