DOI��10.19476/j.ysxb.1004.0609.2017.05.012
Lu-F������ZnO������ʵĵ�һ��ԭ������
������1, 2��������1, 2��֣���� 1, 2
(1. �ӱ���ѧ ������Ϣ����ѧԺ������ 071002��
2. �ӱ���ѧ �������������ģ���о����ģ����� 071002)
ժ Ҫ�����û����ܶȷ�������(DFT)�ĵ�һ��ԭ��ƽ�沨��������(PWPP)������ģ�����δ���ӣ�Lu��F�����Ӽ�Lu-F������ZnO�ļ��νṹ���ܴ��ṹ��̬�ܶȷֲ����������ͽ�糣�������ʡ�������������Ӻ�ZnO�ľ������������䣬����������������Ȳ�ͬ�̶ȵؼ�С���ڹ�ѧ���ʷ��棬F�����ӽ�Lu�����Ӻ�Lu-F������ʱ�ڿɼ�����������ϵ���ͷ���ͣ���ӳǰ���ڿɼ��ⷶΧ���нϸߵ����ʡ�
�ؼ��ʣ�ZnO����һ��ԭ���������ӣ����ӽṹ����ѧ����
���±�ţ�1004-0609(2017)-05-0960-07���� ��ͼ����ţ�0471��0472���� ���ױ�־�룺A
�������������İ뵼��������ѧ��ѧ�������������˹㷺��ע��ZnO��Ϊһ�����͢�-���������(3.37 eV)�뵼�壬��Ϊ����иߴ�60 meV�ļ��������ܺ�ֱ�Ӵ�϶���������ʣ���̫����ء������̽�������������������Լ����崫�����������Ź�����Ӧ��ǰ��[1-4]�����DZ���ZnO������Ҳ����ȱ�ݣ����Խ��������ǿ�ʼͨ������������ZnO�����ʡ����౨��������ϡ��Ԫ����Ϊ���ж��ص��������Ӳ�ṹ����ΪZnO������ϵ���о��ȵ㡣������ZnO�в���ϡ��Ԫ�ؿ���ʵ��ϡ�����ӵĸ�Ч���·��⣬ʵ��1.54 ��m���εĽ����ⷢ�䣬���ZnO������ѹ������[5]����ʵ���ϣ��쾮����[6]�������ܽ����Ʊ��˲�ͬŨ��La���ӵ�ZnO��Ĥ�����ⷢ������Ų���Ũ�ȵ����������ǿ������̲������ƶ��������ص�[7]����ƽ�沨�������Ʒ�������Er��Gd���Ӷ�ZnO���ӽṹ�����յ�Ӱ�죬����ϡ��ԭ�ӵ�4f���о���ӵ��·����ܼ����������ܼ��ij��֣�����鲿�����������죬������ϵ������ϵ����ߡ�CHE��[8]�о����ܽ��������Ʊ���Eu����ZnO��Ĥ���������б�Ĥ�ڿɼ����������ʾ���80%���ϣ�������������������ʽ��ͣ�Eu3+ �����������ܶ����£�δ�ı�ZnO�Ĵ�϶����;�����ۼ��㷽�棬������[9]��Yb2+��Yb3+�ֱ����ZnO����������뱾��ZnO��ȣ������ֲ��Ӷ������˾�̬��纯�������մ��ߺ��ƣ�����0.91eV�������˽�ǿ�����շ塣����ϲ��[10]�о���Y(La)��λZn����ZnO,���������������ӵ�d-d���ԾǨ������µ����շ壬�ı��˽�糣������Ϊ�����µĽ����Ϻ���������ṩ���������ݡ�
Laϵϡ������ZnO���о���������ʵ�黹�����ۼ��㶼�Ѿ��кܶ����ױ�����������LuԪ�ز���ZnO������о����ٱ�������Lu-F�����������Ͽ��Ըı䱡Ĥ�в���������ZnO�����ĵ��Ӻ������Ĵ��ݷ�ʽ�����������Ѿ������Ga-F������ZnO����֤��F���Ӻܴ�̶������ZnO�����ʣ��ڴ˽���һ���о�Lu-F��ZnO���ʵ�Ӱ�졣��һ��ԭ����Ԥ��뵼����Ӻ���������[11-12]���ڲ��Ͻṹ��ơ���������ȷ���õ��㷺��Ӧ��[10]����ˣ������������û����ܶȷ������۵ĵ�һ��ԭ����Lu��F�������Լ�Lu-F�����ӵ�ZnO���м��㲢�Լ��������з���������Ϊʵ���ʵ��Ӧ���ṩ������ۻ�����
1 ����ģ�ͺͷ���
1.1 ����ģ��
�����ZnO��������п��ṹ�������ռ�ȺΪP63mc���Գ���ΪC6v-4���侧����O�����Ƕѻ���Zn�������ܶѻ���c�᷽����Ƕ���ɡ���������ʵ��ֵΪa=b=3.249 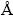 ��c=5.206 ��Oԭ��λ��4������Znԭ���γɵ��������϶�У����γ�O-Zn4 �����壬Znԭ�ӵ����нṹ��Oԭ�����ơ�ѡȡ2��2��2��ZnO�������ṹ���м��㣬�ܹ�32��ԭ�ӣ��ֱ���Fԭ�����Oԭ�ӣ�Luԭ�����Znԭ�ӣ�������Zn15LuO15��Zn16FO15��Zn15LuFO15�ij���������ͼ1��ʾ�����Ӻ�Lu��F�IJ���Ũ��Ϊ3.25%(Ħ������)���ȽϷ���ʵ�ʲ��ӡ�
��c=5.206 ��Oԭ��λ��4������Znԭ���γɵ��������϶�У����γ�O-Zn4 �����壬Znԭ�ӵ����нṹ��Oԭ�����ơ�ѡȡ2��2��2��ZnO�������ṹ���м��㣬�ܹ�32��ԭ�ӣ��ֱ���Fԭ�����Oԭ�ӣ�Luԭ�����Znԭ�ӣ�������Zn15LuO15��Zn16FO15��Zn15LuFO15�ij���������ͼ1��ʾ�����Ӻ�Lu��F�IJ���Ũ��Ϊ3.25%(Ħ������)���ȽϷ���ʵ�ʲ��ӡ�
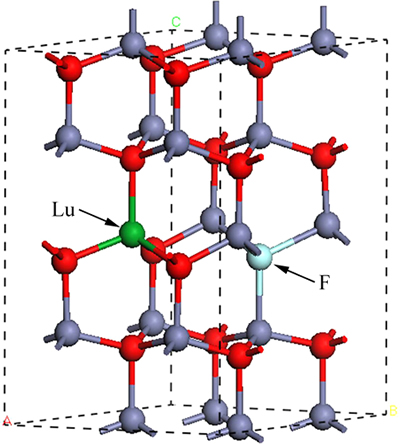
ͼ1 Lu-F������ZnO�ṹģ��ͼ
Fig. 1 Structure model of Lu-F codoped ZnO
1.2 ���㷽��
�ڶ�Lu-F����ZnO��ϵ����ʱ������MS7.0�����е�CASTEP������(Cambridge Sequential Total Energy Package) [13]�����������ȶ�ZnO�������ṹ�����Ż����Ż��е��Ӽ�����õĽ����������ɹ����ݶȽ����µ�PBE��������[14]�����м�����ڵ��ռ��н��С�������ز����������£�����������Ϊ1��10-5 eV/atom��ƽ���ֹ��Ϊ350eV����һ����Ԩ����3��3��1�ָ��ٸ���Ҷ�任����40��40��40�����λ��Ϊ0.001 ��ԭ�Ӽ����������������0.3 eV/nm���������ļ۵���Zn��3d104s2��F��2s22p4 ��Lu��4f145p65d16s2��O��2s22p4��
2 �����������
2.1 �����Ż����
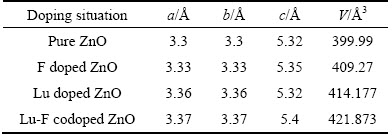
��1����Ϊ��ͬ���������ZnO�ľ������ͳ������������δ���ӵ�ZnO��ȸ���������ϵ��ZnO��������������в�ͬ�̶ȵı仯�����б���ZnO�ļ���ֵ��ʵ��ֵ���������ۼ���ֵ�ӽ�[15]��F-(0.133 nm)�����Ӱ뾶��O2-(0.14 nm)�����Ӱ뾶С�����F��λOԭ�Ӻ�F��Zn����(0.219 nm)����ԭ����O��Zn����(0.200 nm)������F���Ӻ��������䣬���������Lu���ӵ�ZnO��Lu�İ뾶����Zn��ԭ�Ӱ뾶������Lu��O��ƽ����������֮ǰ��Zn��O���������ߵĹ�ͬ���õ��¾������;����������Lu-F�����ӵ�ZnOͬ������������ԭ������¾������ͣ������������
��1 ����ǰ��ZnO���νṹ����
Table 1 Structural parameters of pure and doped ZnO
2.2 �ܴ��ṹ
ͼ2��ʾΪ����ǰ��ZnO�ص�һ����Ԩ���߶ԳƷ�����ܴ��ṹͼ���ܴ�ͼ2��ʼ�ս������ܹ���������ܼ���Ϊ������㡣��ͼ2(a)���Կ���������ZnO�����ͼ۴���λ�ڲ���Ԩ��ͬһ�㼴G�㣬Ϊֱ�Ӵ�϶�뵼�壬��������Ϊ0.719 eV������ξ�����[16]����Ľ���ӽ�������ʵ��ֵ�ͺܶ࣬��������GGA���ƴ�����Ӱ�졣������Ϊһ����Ч�Ľ��Ʒ����������������ֵ�Ƿdz�ȷ�ģ���Ӱ��Լ������Ķ��Է�������ͼ2(b)���Կ���F���Ӻ�����2.405 eV���۴�����2.234 eV�����½������ȼ�С�����ҵ����ͼ۴����ܼ����ܣ������ܼ����뵼��������n�Ͳ��ӡ�ͼ2(c)��ʾΪLu���Ӻ��ZnO���ܴ�ͼ����ͼ2(c)���Կ������Ӻ�ı仯������F���Ӻ�ı仯���ƣ�ʹ�ý������Ƚ�һ����С����СΪ0.633 eV��Lu-F�����Ӻ������������ϵ���������ȱ����С0.363 eV���������ȵļ�С������ʹ�õ��Ӿ��н�С�������Ϳ��Է���ԾǨ�����ҵ����ͼ۴����ܼ���ĸ����ܼ���������ΪLu-F�����Ӿ���ЭͬЧӦ������ڵ����Ӿ��и���ļ�̬��
2.3 ̬�ܶ�
ͼ3��ʾΪZnO����ǰ�������ϵ��̬�ܶ�ͼ����ͼ3(a)�п��Կ���������ZnO�ĵ�����Ҫ��Zn��4s��������ṩ����-6~0 eV��Χ�ڵļ۴���Zn3d��O2p��������ӻ����ɣ���-18~-16 eV�ķ�Χ�ڴ��ڹ�����̬�ܶȷ壬��Ҫ��O2s����ṩ�����Ӻ��ܵ�̬�ܶ���������ܶ��ƶ�������3����̬�ܶȷ��չ���������Լ�����F�����Ӻ�F��2p�����Zn3d�����O2p�����ͬ����-9~0 eV�ļ۴����Ե�������û�й��ף���F2s���ֻ�Ե��ܶ˵ļ۴��й��ף���Բ��Ӳ��ϵ���������Ӱ���С�����Ժ��Բ��ơ�Lu�����Ӻ��Կ����ܵ�̬�ܶ���-5~-4 eV������һ����壬�۲�Lu�ķ�̬�ܶ�ͼ��֪�����̬�ܶȼ������Lu��4f������ף�����Lu5d����Ե���Ҳ�нϴ�Ĺ��ס�Lu-F�����Ӻ��ܵ�̬�ܶ�ͼ�����δ���Ӻ�����������ϵ���Ե�����ܶ��ƶ�(4 eV)�������ܼ����뵼����
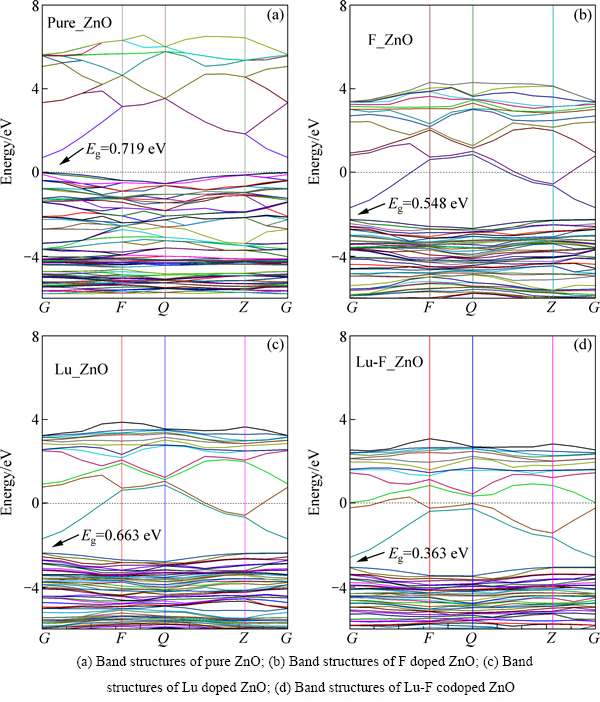
ͼ2 δ���ӺͲ��Ӻ��ZnO�����ܴ��ṹͼ
Fig. 2 Band structures of pure ZnO and doped ZnO
2.4 ��ѧ����
�ڵ�һ��ԭ���о���ͨ���ø���纯������������ĺ�۹�ѧ��Ӧ����������纯����(��)�ı���ʽΪ
��(��)=��1(��)+i��2(��) (1)
ʽ�У���ΪƵ�ʣ���1(��)�ͦ�2(��)�ֱ�Ϊ��纯����ʵ�����鲿����纯�����鲿��2(��)�����ѧ����ֱ��������ģ����Ը��ݵ���ԾǨ��ѡ������ռ��̬��δռ��̬�ĵ��Ӳ�����֮��ľ���Ԫ����õ���ʵ����1(��)��������Kramaer-Kronig��ϵ���鲿��2(��)����õ����ɦ�1(��)�ͦ�2(��)���Լ���������Ĺ�ѧ����[17]��
ͼ4(a)��ʾΪ����ǰ��ZnO�Ľ�纯��ʵ������������仯ͼ����ͼ4(a)�п��Կ�����������ZnO��ʵ����Ҫ��3����ֵ���ֱ������1.7 eV��6.4 eV��10.2 eV�����������ļ�������Ϊ�ӽ�����ϱ���ZnO���ܴ�ͼ��̬�ܶ�ͼ��֪��1.7 eV���ҵķ�ֵ��Ҫ��O2p��Zn4s���֮��ĵ���ԾǨ�γɣ�6.4 eV���ҵķ�ֵ��Ҫ��Zn3d��O��2p���֮��ĵ���ԾǨ�γɣ�10.2 eV���ҵķ�ֵ��Ҫ��O2s��Zn3d���֮��ĵ���ԾǨ�γɡ����Ӻ�Խ�纯����Ӱ����Ҫ�����ڵ��ܶˣ�F�����Ӻ��һ������ʧ��������ΪF���Ӻ��ܴ����ƣ���1.7 eV���Ҳ�����O2p��Zn4s���֮��ĵ���ԾǨ����ģ�������0~4 eV��Χ����������ܶ��ƶ���Lu�����Ӻ�ͬ����һ����Ҳ��ʧ����Ҳ��������ԭ������ģ�������6.4 eV�����γ�һ�����ڱ���ZnO�ķ�ֵ��������ΪLu��4f�������λ��4.8 eV�����������ڲ�ͬ���ܼ�֮�����ԾǨ����̬�ܶ�ͼ��Ҳ���Կ�����4.4 eV����Lu4f�����̬�ܶ��нϴ�Ĺ��ף�������6.4 eV���һ���Zn3d�����O2p������ӵ�ԾǨ�������γ�һ���Ƚϴ�ķ�ֵ��Lu-F�����Ӻ��ڸ��ܶ˵ı仯������Lu���������ƣ������ڵ��ܶ�0.5 eV�����γ�һ����ֵ�����һ���һ����С�IJ��ȣ�����Lu-F�����Ӳ�����Ӱ�졣
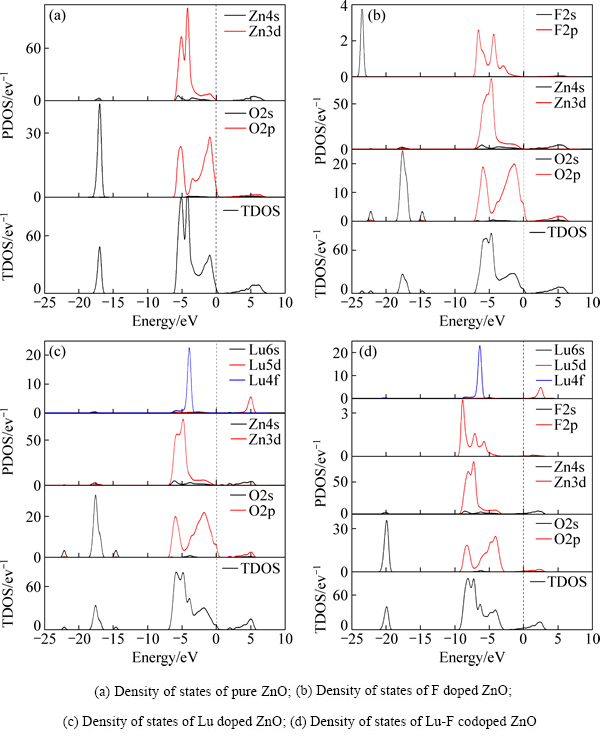
ͼ3 δ���ӺͲ��Ӻ��ZnO������̬�ܶ�(DOS)�ͷ�̬�ܶ�(PDOS)ͼ
Fig. 3 Density of states(DOS) and partial density of states(PDOS) of pure ZnO and doped ZnO
ͼ4(b)��ʾ�Dz���ǰ��ZnO������ϵ��ͼ���ⲿ�ּ���Ϊ��ʹ����ʵ��ֵ����������˼���������������ֵȡ2.6 eV(�������ȵ�ʵ��ֵ�����ֵ֮��)����ͼ�п��Կ�����������ZnO���մ���λ��2.9 eV���ӽ��ڴ�϶��ʵ��ֵ3.37 eV��������3������ֵ���ֱ�λ��4.5 eV��10.2 eV��13.8 eV�����������[18]��������Ϊ�ӽ���F��Lu�����Ӻ��ڵ��ܶ����մ��߾�����ܶ˷����ƶ�����ͬ����Lu�����Ӻ���12 eV���Ҳ���һ���ϸߵķ�ֵ����˵��Lu���Ӻ�������ZnO������ⷶΧ�ڵ�����ϵ������Lu-F�����Ӻ��ڸ��ܶ˵ı仯��Lu�����ӱ仯���ƣ��ڵ��ܶ˱���Ϊ�����մ��ߵĺ��ƣ������˶Կɼ�������շ�Χ��ͼ4(c)��ʾΪZnO����ǰ�����ʱ仯������ڿɼ��ⷶΧ�ڣ�������ZnO�ķ����λ��14.8 eV���ң����ӵ��·���巢����ͬ�̶ȵ�����ܷ����ƶ������й�����ʱ������ƶ���Ϊ���ԡ�Lu-F�����Ӻ�Lu����ʱ������������ԣ���F������ʱ�����仯��С��˵��ǰ��������������ӡ�������ʧ���Է�ӳ������ͨ�����ȵĵ����ʱ��������ʧ�����ͼ4(d)��ʾΪZnO����ǰ��������ʧ��������ZnO������ʧ��ֵλ��15 eV���ң�����ʵ��ֵ18.8 eVʮ�ֽӽ�[19]�����Ӻ�������ʧ���ֲ�ͬ�̶ȵĺ��ƣ�����Lu-F������ʱ���������ԡ�ֵ��ע����ǣ�������ʧ��ֵ�뷴�����ߵ�Ѹ���½�ֱ�Ӷ�Ӧ��
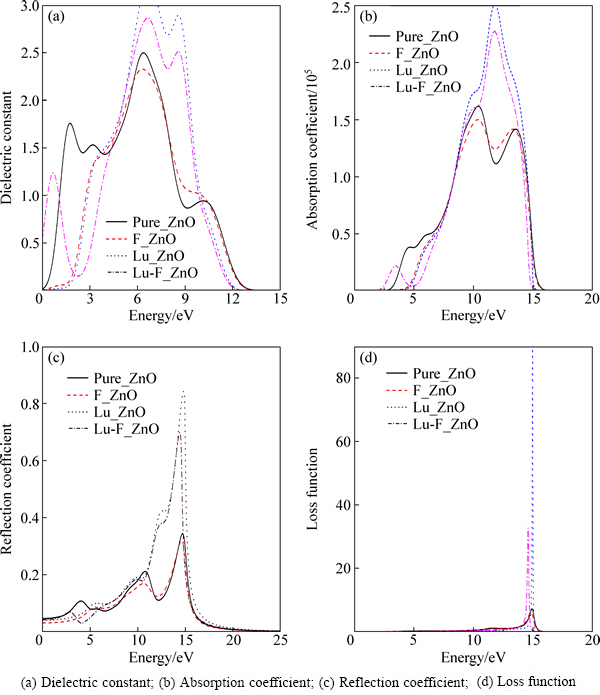
ͼ4 δ���ӺͲ��Ӻ��ZnO��ѧͼ��
Fig. 4 Optical patterns of pure ZnO and doped ZnO
3 ����
1) ���ӵ��¾�����䣬������ͣ����Ӻ������ǰ��ZnO��Ƚ������ȼ�С�������ͼ۴����ܼ����ܣ������ڵ����ڴ�϶���ԾǨ��
2) ���Ӻ�ZnO�Ĺ�ѧ���ʵõ����ƣ�����Lu���Ӻ�����ϡ��Ԫ������������ӽṹ�������˵��ӵ�ԾǨ���ʣ�������ǿ���������������ϵ���Ϳɼ�������շ�Χ��
��л����л�ӱ���ѧ����ͤ����Ϊ�����ṩCASTEP��������������������ۡ�
REFERENCES
[1] LEE K E, WANG M, KIM E J, HAHN S H. Structural, electrical and optical properties of sol�Cgel AZO thin films[J]. Current Applied Physics, 2009, 9(3): 683-687.
[2] HASABELDAIM E, NTWAEABORWA O M, KROON R E, SWART H C. Surface analysis and cathodoluminescence degradation of undoped ZnO and ZnO: Zn phosphors[J]. Journal of Vacuum Science & Technology B, 2016, 34(4): 041221(1-8).
[3] LANG Ji-hui, HAN Qiang, YANG Jing-hai, LI Chang-sheng, LI Xue, YANF Li-li, ZHANG Yong-jun, GAO Ming, WANG Dan-dan, CAO Jian. Fabrication and optical properties of Ce-doped ZnO nanorods[J]. Journal of Applied Physics, 2010, 107(7): 074302(1-4).
[4] GORAI P, ERTEKIN E, SEEBAUER E G. Surface-assisted defect engineering of point defects in ZnO[J]. Applied Physics Letters, 2016, 108(24): 241603(1-5).
[5] ������, ������, �� ��. ϡ������ZnO��Ĥ���о���չ[J]. ���ϵ���A, 2012, 26(8): 32-37.
LI Ying-juan, CHEN Qing-ming, MA Ji. Research progress of rare earth doped ZnO thin films[J]. Materials Review, 2012, 26(8): 32-37.
[6] �쾮��, ���г�. ��ͬŨ��La����ZnO��Ĥ��ѧ���Ե��о�[J]. ͨ��ʦ��ѧԺ��, 2010, 31(4): 24-25.
XU Jing-hua, LIU You-cheng. The study of La doped ZnO thin films optical properties[J]. Journal of Tonghua Normal University, 2010, 31(4): 24-25.
[7] ������, �� ��, ��Ӣ��, �ƽ���, �� Ӣ, ����ϲ. ��һ��ԭ���о�ϡ������ZnO�ṹ�Ĺ������[J]. ����ѧ��, 2013, 62(4): 047101(1-6).
LI Hong-lin, ZHANG Zhong,  Ying-bo, HUANG Jin-zhao, ZHANG Ying, LIU Ru-xi. First principles study on the electronic and optional properties of ZnO doped with rare earth[J]. Acta Physica Sinica, 2013, 62(4): 047101(1-6).
Ying-bo, HUANG Jin-zhao, ZHANG Ying, LIU Ru-xi. First principles study on the electronic and optional properties of ZnO doped with rare earth[J]. Acta Physica Sinica, 2013, 62(4): 047101(1-6).
[8] CHE P, MENG J, REN L R, LIN G. Fabrication and magnetic properties of highly oriented ZnO:Eu films by sol-gel process[J]. Journal of Rare Earths, 2006, 24(1): 298-301.
[9] �����, �����, ������, �� ��. ��ͬ��̬ϡ��Ԫ��Yb����ZnO�ĵ��ӽṹ��ѧ����[J]. ����ѧ��, 2013, 62(12): 127101(1-7).
LIU Wei-jie, SUN Zeng-hao, HUANG Yu-xin, LENG Jing. Electronic structures and optical properties of rare earth element(Yb) with different valences doped in ZnO[J]. Acta Physica Sinica, 2013, 62(12): 127101(1-7).
[10] ����ϲ, ������, ������, ������, �� ��, �� �. ϡ��Ԫ��(Y, La)����ZnO�ĵ��ӽṹ��ѧ����[J]. ����ѧ��, 2011, 60(1): 017101(1-7).
WU Yu-xi, HU Zhi-xiang, GU Shu-lin, QU Li-cheng, LI Teng, ZHANG Hao. Electronic structures and optical properties of rare earth element (Y, La) doped in ZnO[J]. Acta Physica Sinica, 2011, 60(1): 017101(1-7).
[11] �ι���, ��˼��, κ����, ������, �� �. HfxTi1-xO2���ӽṹ���ѧ���ʵĵ�һ��ԭ���о�[J]. ��������ײ�ѧ��, 2010, 29(4): 264-267.
DUAN Guo-yu, SONG Si-chao, WEI Chang-dong, WANG Song-you, JIA Yu. Electronic structures and optical properties of HfxTi1-xO2 calculated from first peinciples[J]. J Infrared Millim Waves, 2010, 29(4): 264-267.
[12] �Ż���, �ϻ���, �� ��. Ga/N�����Ӷ�InSb���ӽṹ��Ӱ��[J]. ��������ײ�ѧ��, 2012, 31(3): 231-234.
ZHANG Hui-yuan, XING Huai-zhong, ZHANG Lei. The effect of Ga/N Co-doping on electronic structure og InSb[J]. J Infrared Millim Waves, 2012, 31(3): 231-234.
[13] CLARK S J, SEGALL M D, PICKARD C J, HASNIP P J, PROBERT M J, REFSON K, PAYNE M C. First principles methods using CASTEP[J]. Z Kristallogr, 2005, 220(5/6): 567-570.
[14] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77: 3865-3868.
[15] �� ��, ���㺭, �� ��. Mn����ZnO��ѧ���Եĵ�һ��ԭ������[J]. ����ѧ��, 2008, 57(2): 1054-1060.
CHEN Kun, FAN Guang-han, ZHANG Yong. First principles study of optical properties of wurtzite ZnO with Mn-doping[J]. Acta Physica Sinica, 2008, 57(2): 1054-1060.
[16] �ξ���, ֣����, ������, ʷ��ٻ, ��С��. Cu-Co������ZnO������ʵĵ�һ��ԭ������[J]. ����ѧ��, 2014, 63(4): 46301(1-9).
HE Jing-fang, ZHENG Shu-kai, ZHOU Peng-li, SHI Qian-ru, YAN Xiao-bing. First-principles calculations on the electronic and optical properties of ZnO codoped with Cu-Co[J]. Acta Physica Sinica, 2014, 63(4): 46301(1-9).
[17] ��ѧ��. �뵼�����ѧ����[M]. 2��. ����: ��ѧ������, 2002: 23-24.
SHEN Xue-chu. Spectroscopy and optical properties of semiconductors[M]. 2nd ed. Beijing: Science Press, 2002, 23-24.
[18] �� ��, �� ǿ, ����ѫ, ������, �� ��, ������, �� ��, ����ͤ. Al��Ni������ZnO��ѧ���ʵĵ�һ��ԭ���о�[J]. ����ѧ��, 2009, 58(8): 5624-5630.
GUAN Li, LI Qiang, ZHAO Qing-xun, GUO Jian-xin, ZHOU Yang, JIN Li-tao, GENG Bo, LIU Bao-ting. First-principles study of the optical properties of ZnO doped with Al, Ni[J]. Acta Physica Sinica, 2009, 58(8): 5624-5630.
[19] LU J G, FUJITA S, KAWAHARAMURA T, NISHINAKA H, KAMADA Y, OHSHIMA T, YE Z Z, ZENG Y J, ZHANG Y Z, ZHU L P, HE H P, ZHAO B H. Carrier concentration dependence of band gap shift in n-type ZnO: Al films[J]. Journal of Applied Physics, 2007, 101(8): 83705(1-7).
First-principles calculations on electronic and optical properties of ZnO codoped with Lu-F
ZHANG Ming-ju1, 2, LI Wen-ming1, 2, ZHENG Shu-kai1, 2
(1. College of Electronic and Informational Engineering, Hebei University, Baoding 071002, China;
2. Research Center for Computational Materials and Device Simulations, Hebei University, Baoding 071002, China)
Abstract: The geometry and band structures, density of states, light absorption spectra and dielectric constants of pure ZnO, Lu, F single doped, and Lu-F co-doped ZnO were calculated using the plane-wave ultra-oft pseudo-potential (PWPP) method based on density functional theory (DFT). The calculated results indicate that the lattice constants of doped ZnO are distorted and the volumes increase. The band gaps of the doped ZnO are reduced with different degrees. In terms of the optical properties, the absorption coefficient and reflectivity of F-ZnO are smaller than those of Lu-ZnO and (Lu-F)-ZnO, which indicate a higher transmittance of the F doped ZnO in the visible light range.
Key words: ZnO; first principle; codoped; electronic structure; optical property
Foundation item: Project(ZD2017008) supported by Key Program of Science and Technology Research of Colleges and Universities in Hebei Province, China
Received date: 2016-04-21; Accepted date: 2016-10-20
Corresponding author: ZHENG Shu-kai; Tel: +86-15932188935; E-mail: zhshk@126.com
(�༭ �� ��)
������Ŀ���ӱ�ʡ�ߵ�ѧУ��ѧ�����о��ص���Ŀ(ZD2017008)
�ո����ڣ�2016-04-21�������ڣ�2016-10-20
ͨ�����ߣ�֣�����������ڣ���ʿ���绰��15932188935��E-mail��zhshk@126.com

