���±�ţ�1004-0609(2008)09-1692-07
LaNi5(111)����ṹ����������ĵ�һ��ԭ���о�
�����£�֣��ɽ���� ������ ��������� ��
(������ѧ ������ѧ�빤�̼���ѧԺ����������ɫ�������ϼ���ӹ��¼����ص�ʵ���ң����� 530004)
ժ Ҫ�����û����ܶȷ������۵ĵ�һԭ������ƽ�沨������������Ͻ�LaNi5��LaNi5(111)����ĵ��ӽṹ���м��㣬��Hԭ����LaNi5(111)��������ģ�ͽ��й����Ż������������LaNi5(111)�����ԥ�ṹLaԭ����������Niԭ����������������ƽ�ı�������ӱ���ԭ����Hԭ�ӵĽӴ��������������Ч���Լ����2.3%��������Hԭ�����������ɢ��������о����0.5�����ӣ������ڱ�����ϵĵ���ת�Ƶ�Hԭ���ϣ�H2���ӽ��������Hԭ�Ӻ���LaNi5(111)�����ƽ���ȶ��ṹ���⻯��LaNi5H7������ͬλ�õĽṹ��Ϊ���ƣ�����H2������LaNi5(111)����Ľ��������������䷴Ӧ���ԼΪ0.27 eV��
�ؼ��ʣ�LaNi5�����ӽṹ�������������һ��ԭ��
��ͼ����ţ�TG 239.7���� ���ױ�ʶ�룺A
First principle investigations on surface structure and
mechanism of hydrogen adsorption of LaNi5(111)
LIU Yi-xin, ZHENG Ding-shan, ZHANG Yi, JIANG Long, LI Guang-xu, GUO Jin
(Key Laboratory of National Education Ministry for Nonferrous Metals and Materials Processing Technology,
College of Physics Science and Technology, Guangxi University, Nanning 530004, China)
Abstract: The electronic structures of LaNi5 hydrogen storage alloy and LaNi5(111) surface with hydrogen atoms were calculated by plane wave pseudo-potential method based on density functional theory. The results show that on the relaxed surface, La atoms protrude from surface and Ni atoms cave in, which enlarges the contacting area with H atoms. The effective volume of the surface layer is increased by 2.3%, which favors H atoms to diffuse into bulk from the surface. Calculated charge population presents negative charge on the surface, and the negative charge may transfer from the surface layer to H atoms. The stable structure by geometry optimizing after H2 molecule is dissociated into two H atoms on LaNi5(111) surface presents similar structure with hydride LaNi5H7 at the same position. The possible dissociation path and the mechanism of hydrogen-adsorbed are investigated with transition state method, and the activation energy of reaction is estimated as 0.27 eV.
Key words: LaNi5; electronic structure; hydrogen adsorption mechanism; first principle
���������Ϊһ�������ܲ��ϣ����������ǵ��ձ��ע���о�������ϵ��۽ṹ�Լ�������״̬�������˽����������ϵ����ܣ�Ѱ���µ��������������Ҫ�����塣AB5�ͻ��ϡ���Ͻ���Ϊ��������ѵõ��㷺ʹ�ã�LaNi5��AB5�ͻ������н�Ϊ����Ķ�Ԫ������ϣ�����ΪNi/MH��صĸ�������[1]����ʵ������������۽ṹ���õ���Ϊȫ����� ��[2?6]��Ȼ�����ڱ���ʵ����������Ա���ԭ�ӽṹ�������ԥ������ɡ��Լ��������ܵȷ�����о�, �����ǶԸ��ӵĺϽ��������о�����Ȼȱ����ֵ�ʵ�����ݡ���ˣ�ͨ�����ۼ����о��Ͻ�����ԭ������ӽṹ�ѳ�Ϊһ����Ҫ�ľ�������о���������LaNi5�Ͻ���������У�������LaNi5�����е�H����ԭ��״̬���ڵģ���LaNi5���棬H2��������Ҫ�����Hԭ�Ӻ��ٽ��뾧�塣
�ڵ�һԭ�������У����õļ���ģ�������֣����Ŵ�ģ�ͺͳ�����ģ�͡��Ŵ�ģ������ģ������ԭ�ӵĹ���ϵͳ����������ģ��ģ����Ǿ���һ���Գ��Ա߽���������ά������ϵͳ�����Գ�����ģ�ͱ��Ŵ�ģ���ܷ�Ӧ����״̬����ʵ�ľ���ϵͳ���������߲��û����ܶȷ�������(DFT)�ĵ�һԭ������ƽ�沨(PW-PP)[7]���������㲢ģ�ⲻͬ��ʼ״̬��H2���ӽ��������Hԭ�Ӻ�Hԭ����LaNi5(111)���ϵ��������̣���H/LaNi5(111)��ϵ�Ĺ����Լ����ӽṹ�����о���
1 ���㷽��
ʹ��CASTEP����[8]���м��㣬�ȶ�LaNi5�������м����Ż����ټ����䵥���ܡ������У�����2��1������ģ�ͣ�ѡ����ڹ����ݶȽ���(GGA)�µ�PBE[9]����������������Vxc��ƽ�沨��ֹ��ȡΪ300 eV��Brillouin����k���ȡΪ6��6��6�������ڵ��ռ��Ͻ��У�ģ�Ͱ��������ͶԳ���(P1)�����Ż������������е���������Ϊ1��10?5 eV/atom����������ÿ��ԭ���ϵ���������0.003 eV/nm����Ӧ�������� 0.05 GPa�������������ģ�ͣ���ԭ�ӵ���������̬�ֱ�Ϊ��La 4s24p64d104f05s25p65d16s26p0��Ni 3s23p63d84s24p0��H 1s1��
Hԭ����LaNi5(111)����Ľ����?EΪ����ǰ�����������IJ�ֵ���ù�ʽ����Ϊ
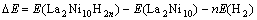
ʽ�� E(La2Ni10H2n)Ϊ������������������E(La2Ni10)Ϊ����ǰ����������������E(H2)ΪH2���ӵ�������
2 ������������
2.1 �ṹ�Ż�
LaNi5��һ�־���CaCu5�;���ṹ��ϡ������Ͻ𣬿ռ�ȺΪP6/mmm������Laԭ��ռ��laλ��ԭ������Ϊ(0, 0, 0)��Niԭ��ռ������λ�ã�����z=0�Ļ�ƽ���2cλ�ʹ���z = 1/2���м�ƽ���3gλ��2cλԭ������Ϊ(1/3, 2/3, 0)��(2/3, 1/3, 0)��3gλԭ������Ϊ(1/2, 0, 1/2)��(0, 1/2, 1/2) ��(1/2, 1/2, 1/2)����ṹ��ͼ1��ʾ�����ȶ�LaNi5����ṹģ�Ͱ��������ͶԳ���(P1)�����Ż����Ż�������ڱ�1���Ż�����Ҫ��a��b������c��仯�dz�С��c/aֵ��ʵ��ֵ�ȽϽӽ��������Լ0.98%����������LaNi5����Ͻ���ڸ������ԡ��ӱ�1�л��ɿ����Ż���ľ�������������ӣ���ʵ��ֵ����Լ2.1%��
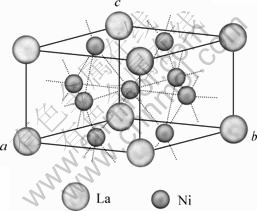
ͼ1 LaNi5����ģ��
Fig.1 Crystal model of LaNi5
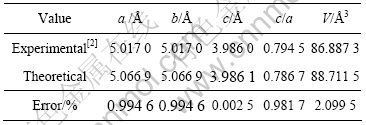
��1 LaNi5���徧��������������ʵ��ֵ�Ƚ�
Table 1 Comparison of theoretical and experimental values of lattice parameters of LaNi5 crystal
2.2 LaNi5 (111)����ij�ԥ��������ӽṹ
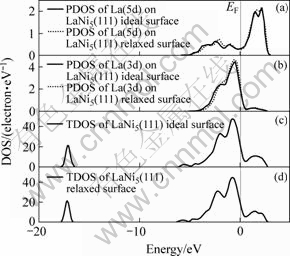
����LaNi5(111)���ܶȽϸߣ������о�H��LaNi5(111)��������ʱ��ת����̱����������ϸ�����һ���ԡ����ǵ����澧����ƽ�ƶԳ��Ժ;ֲ���Ⱥ�Գ��ԣ��Լ�����ԭ�ӵı߽�ЧӦ�������в��õ�LaNi5(111)���澧��ģ�ͼ�ͼ2(a)��ʾ������������2��1�������ɣ���ղ�����Ϊ7 ?���Ա�֤����֮�䲻�������Ե�����á�
���ȿ���LaNi5 (111)������ij�ԥ�������LaNi5 (111)��������й����Ż���������ڱ�2���������ͳ�ԥ����ľ���ģ�ͷֱ���ͼ2(b)��ͼ2(c)��ʾ���ӱ�2���Կ�����������La2ԭ�������ƶ�������Լ0.001 9 nm��������Ni2��Ni4ԭ�������ƶ�0.010 8 nm����Ni8��Ni10ԭ�������ƶ�0.015 1 nm���α����Ni6ԭ�ӷ�����С��λ���ƶ����������ƶ�0.000 3 nm������ڶ���Laԭ�������ƶ��� 0.021 1 nm������ڶ���Ni1��Ni3ԭ�������ƶ� 0.009 8 nm��Ni7��Ni9ԭ��Ҳ�����ƶ�Լ0.014 7 nm����Ȼ������ƽ�ı���������˱���ԭ����Hԭ�ӵĽӴ������������Hԭ�������ԭ�Ӽ������á�����������ڶ���ԭ�Ӽ�����������������������������Laԭ�ӵ����ƺ�Niԭ�ӵ����ƣ����±��澧���������������Լ2.3%���⽫������Hԭ�Ӵ������������������ɢ��
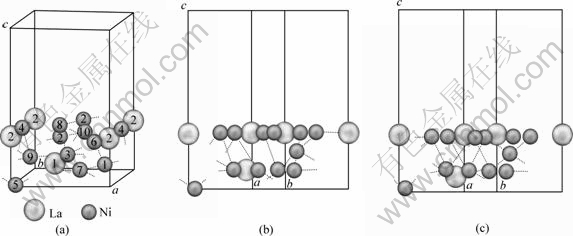
ͼ2 LaNi5(111)���澧��ģ��
Fig.2 Crystal models of LaNi5(111) surface: (a) Surface of LaNi5(111); (b) Side view of ideal surface of LaNi5(111); (c) Side view of relaxed surface of LaNi5(111)
��2 LaNi5(111)��ԥ�����湹���Ż����
Table 2 Geometry optimization parameters of LaNi5(111) relaxed surface
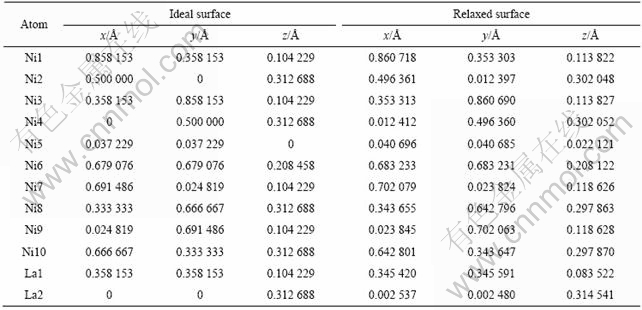
Ϊ�������˽�LaNi5(111)����ṹ��ԭ�Ӽ������ã�����������������ͳ�ԥ���������̬�ܶ�(TDOS)���ֲ�̬�ܶ�(PDOS)ͼ�������ͼ3��ʾ��ͼ�н������ܼ�Ef��ȡΪ��㣬��Ϊ�����IJο��㡣��ͼ3�п��Կ���������ͳ�ԥ����������Ƶ�̬�ܶ�ͼ�������ܼ����������ͳ�ԥ�����̬�ܶ�һ�������ϵ͡����ݽṹ�ȶ���������ܼ�����̬�ܶȹ�ϵ�������ܼ��ϵĵ�̬�ܶ�ֵ��Ӧ���ȶ��ṹ���������ڷ����ܼ�̬�ܶ�ԽС������ζ�Ÿ�����Ӳ���ɼ������ṹ�����ȶ�[10]��LaNi5(111)�������ķ�����λ��Ni(3d)�����ܴ��ĸ��ܶ��±��أ���̬�ܶ�Ѹ���½���λ��Ni(3d)�����ܴ���һ��δ�����ܴ�����������ת�ƵĿ����Խϴ�˵��LaNi5(111)������Ȼ�ǽ����ԣ�������[7]�ļ�����һ�����ڷ�������La��5d���ӹ���С���������������ȣ���ԥ����ķ����ܼ��·���DOS������λ�þ�����ܼ������н�С��Ư��(Լ0.1 eV)��˵���ṹ���ڸ��ȶ���
ͼ3 LaNi5(111)������漰��ԥ������̬�ܶȼ��ֲ�̬�ܶ�ͼ
Fig.3 Total and partial densities of states of LaNi5(111) ideal surface and relaxed surface
LaNi5(111)������ԭ�ӵ�Mulliken��ɷ���������3���С�Laԭ��ʧȥ���ӣ��������ԣ���Niԭ�Ӷ��õ����ӣ��ʸ����ԡ���������ԭ�ӷ����˵��ת�������Ӵ�Laԭ��ת�Ƶ�Niԭ���ϡ������Laԭ�Ӷ��ԣ������һ��La2ԭ��ʧȥ1.30�����ӣ��ڶ���La1ԭ��ʧȥ1.06�����ӡ�������������Niԭ�ӣ������һ���һ��Niԭ��(Ni8��Ni10)�õ�0.24�����ӣ�ÿ���ڶ���Niԭ��(Ni2��Ni4)�õ�0.16�����ӡ������ʧȥ�ĵ��ӱȵõ��ĵ��Ӷ�0.5��������0.5�������ӣ������ڱ����ĵ���ת�Ƶ�Hԭ���ϡ�
��3 LaNi5(111)��LaNi5(111)��2H�����ĸ�ԭ�ӹ����ɷֲ�
Table 3 Charges population on atomic orbits in LaNi5(111) and LaNi5(111)��2H surface layer

2.3 H��LaNi5 (111)������ȶ��ṹ�����ӽṹ
һ����Ϊ���������⻯����������������ѧ�������⻯���γɵȽΣ�����������������ѧ�����Ĺ����У�H2�����ѽ�ΪHԭ�ӣ�����������ѧ������ΪH+����������H+����ʽ��Ͻ��ϳ��⻯�����������̾�����ʱ��̣ܶ�������ʵ���ֶι۲쵽��HAMMER��[11]�ɹ�����������ͷ�Ӧ;���о�H2�����ڽ�����������������̣������ʾH2����ͨ���������;��ֱ�������ȶ�λ�á�
Ϊ���о�LaNi5(111)�����������ȶ�λ�ã����γ��ȶ���ԭ������̬�����ȶ�H2��������Hԭ�Ӻ���LaNi5(111)����Ľṹ���нṹ�Ż���Hԭ����LaNi5(111)������ܵ�λ��������6�֣��ٴ��ڱ�����Niԭ�����Ϸ��Ķ�λT���ڳ���λLB���۶���λSB1���ܶ���λSB2����λ��La1ԭ���Ϸ��Ŀ�λH1����λ��Ni6ԭ���Ϸ��Ŀ�λH2�������ͼ4(a)��ʾ��
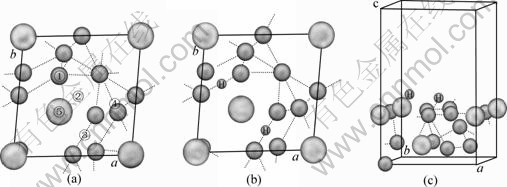
ͼ4 LaNi5(111)��LaNi5(111)��2H����ģ��
Fig.4 Crystal models of LaNi5(111) and LaNi5(111)��2H: (a) Top view of LaNi5(111); (b) Top view of LaNi5(111)��2H; (c) Side view of LaNi5(111)��2H
�������ṹ�Ż������У�����ǰ��IJ������䡣����û�иı�La��Niԭ�ӵ����꣬����Ϊ�������������������С������Ҫ����H2�����������ʱ���ڣ������Hԭ�������ϴ�Ľ���Laԭ�Ӻ�Niԭ������������λ��ƫ�ơ����ȶ�����Hԭ����LaNi5(111)���ϵ�14�ֲ�ͬ��ʼ���ͽ����˽ṹ�Ż����Ż�������ڱ�4�С�
��4 ��LaNi5(111)���ϵ�14�ֲ�ͬ��ʼλ�ý��нṹ�Ż��������������������
Table 4 Energy, bond length and bond angle of optimized structures of 14 kinds of different initial positions of LaNi5(111) surface
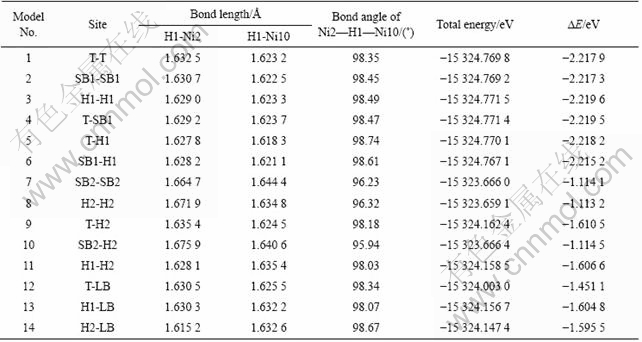
�Ƚ�LaNi5(111)���ϵ�14�ֲ�ͬ��ʼλ�õĽṹ�Ż���������Է��֣�Hԭ�Ӿ�λ��La1ԭ���Ϸ��Ŀ�λH1����ʱ(Model 3)���Ż������������ͣ����ýṹ��Ϊ�ȶ�����λ��Ni6ԭ���Ϸ��Ŀ�λH2����ʱ(Model 8)���Ż������������ߣ������ýṹ��ȶ���Hԭ����λ�ڶ�λT�Ͷ���λSB1ʱ(Model 4)���Ż���Ľ����λ��La1ԭ���Ϸ��Ŀ�λH1����ʱ�Ż��Ľ���dz��������Hԭ�Ӿ�λ�ڶ���λSB2ʱ(Model 7)���Ż���Ľ����λ��Ni6ԭ���Ϸ��Ŀ�λH2����ʱ�Ż��Ľ���dz��������������Ż����ƽ���ȶ��ṹ���жԳ��ԣ���ͼ4(b)��ʾ���ڱ�4��ֻ�г�H1��Ni2��Ni10���������ǣ����⻯��LaNi5H7����ṹͬһλ�ô���H��Ni�ɼ��ļ����ֱ�ԼΪ1.629 2 ?��1.671 3 ?��Ni��H��Ni�ļ���ԼΪ102.82?���ɴ˿��Կ���Hԭ����LaNi5(111)�����ƽ���ȶ��ṹ���⻯��LaNi5H7������ͬλ�õĽṹ��Ϊ���ơ�Hԭ���ڽ���Ͻ��γ��⻯��ʱ������ռ�������ȶ���λ�ã���ͼ4(b)��4(c)��ʾ��
ͼ5��ʾΪLaNi5(111)��2Hƽ���ȶ��ṹ(Model3)����̬�ܶ�(TDOS)���ֲ�̬�ܶ�(PDOS)ͼ����ͼ5��֪��λ�ڷ����ܼ�EF��?34.1~ ?32.6 eV��?17.9~ ?16.2 eV�����ڷֱ���La(5s)��La(5p)����̬�ܶȵĹ��ס������������������?6.9~ ?3.5 eV����ֵ�������ӣ���Ҫ��Ni(3d)���������H�ĵ���̬�ܶ��γɵġ��ڷ����ܼ�EF�����ķ���Ҫ��Ni(3d)����ĵ����ṩ��������λ��Ni(3d)�����ܴ��ĸ��ܶ��±��أ���̬�ܶ�Ѹ���½���λ��Ni(3d)�����ܴ���һ��δ�����ܴ���˵��LaNi5(111)��2H��Ȼ�ǽ����ԡ������������ܼ����ֵ�������Ni(3d)���������������Ҫ����Ni(3d)��La(5d)�����Ni(3p)������ӵĹ���Ҳ���ɺ��ԡ�
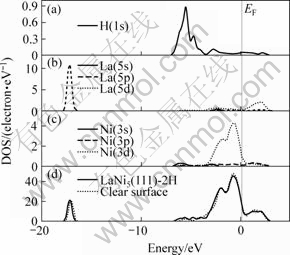
ͼ5 LaNi5(111)��2H����̬�ܶȼ��ֲ�̬�ܶ�ͼ
Fig.5 Total and partial electron densities of states of LaNi5(111)��2H
��3Ҳ�г���ϵ����ƽ��������Laԭ�Ӻ�Niԭ�Ӽ������ڱ����Hԭ�ӵĸ�ԭ�ӹ������ռ�����Լ�����������ɱ�3������ԭ�ӵ�ɷֲ��ɼ�������Hԭ�Ӻ����La2ԭ��ʧȥ���ӣ�Niԭ��Ҳʧȥ���ӣ����Ni��Ni�������������������ÿ��Hԭ�ӵõ�0.25�����ӣ�ʹH ԭ�Ӵ������Եĸ���ɡ�ͼ6��ʾΪLaNi5(111)���漰LaNi5(111)���������ĵȵ���ܶ�ͼ����ͼ�п��Կ�����LaNi5(111)�������Ni2��Ni10֮��ĵ������нϴ���ص����гɼ����á�Hԭ��������LaNi5(111)�����Ni2��H��Ni10��H֮��ĵ������ص��ϴ�����֮��������Ҫǿ��Ni2��Ni10ԭ�Ӽ������ã�Ni2-Ni10֮������������������

ͼ6 LaNi5(111)��LaNi5(111)��2H�ĵȵ���ܶ�ͼ
Fig.6 Contour maps of electron density of LaNi5(111) and LaNi5(111)��2H: (a) LaNi5(111); (b) LaNi5(111)��2H
2.4 ����̬��������ṹ
ͨ�������Ż������õ�Hԭ����LaNi5(111)�����ƽ���ȶ��ṹ��Ϊ�˽�һ���˽�H2��LaNi5(111)����ķֽ�ͷ�Ӧ���̣���������Эͬ�任 (Linear synchronous transit�CLST)�����Эͬ�任(Quadratic synchronous transit�CQST)���ϵķ���[12]�������ݶ�(Conjugate gradient�CCG)����[13?14]�о�����̬(Transition state�CTS)���÷����Ĺؼ������ڶԳ�����������ʹ��һЩ��Ӧ����(��������)���ù̶��ڹ���̬�ϣ���LST/QST�����Ż�����ʹ���Ż��ṹ�뿪
����̬���Ż���������δ�̶������꣬���Ż�Hԭ�ӵ����꣬�Ӷ�����ʹ�÷�ʱ�Ĺ���̬�Ż���������
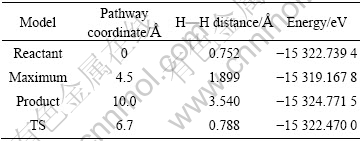
����H2���Ӿ���LaNi5(111)������0.4 nm���ij�ʼλ�ýṹ���ھ��������Ż�����Ϊ��Ӧ��Ż����õ���ƽ���ȶ��ṹ��Ϊ���뷴Ӧ�IJ����5����Ϊ��Ӧ�����и�̬�Ĺ��Ͳ������������ӷ�Ӧ��㾭������̬�õ���������Ӧ·������Ӧ�Ľṹ��ͼ7��ʾ��
��5 ��Ӧ�����и��м�̬�Ĺ��Ͳ���������
Table 5 Geometry parameters and energy of each image for reaction
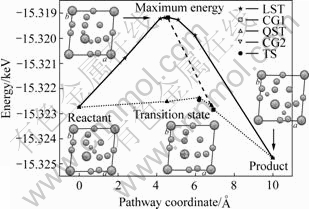
ͼ7 ��Ѱ����̬ʱ�ط�Ӧ·���������仯����
Fig.7 Variation of total potential energies along reaction pathway during transition state search
��ͼ�п��Կ�������ͨ��LST/QST�����Ż��õ�����̬���о��У��ӷ�Ӧ�↑ʼ���������ӵ����ֵ�����õ���������ṹ����ʱ��H��H����ԼΪ 0.189 9 nm��H2�����ѽ����Hԭ�ӡ���ʵ���������������ṹ���ܲ�������������Ѱ�Ĺ���̬�ṹ����������ִ����ֵ��С����������������˺�LST/QST����������Ѱ���ҵ�����̬������̬λ����ͼ7����ʾ��
���������仯��������������ṹ�뷴Ӧ��֮��������������ӷ�Ӧ�ﵽ����������˷����������ԼΪ3.57 eV����Ҳ��H2������LaNi5(111)��������Hԭ��������������������Ѱ����̬�ķ�Ӧ·���У����������;���ڹ���̬����(TS)����һ�����ݣ���H2������LaNi5(111)����Ļ���ݣ����ɹ���̬�뷴Ӧ�������֮�����ó�����ֵԼΪ0.27 eV����ʱH2���Ӽ��Ϊ0.078 8 nm��Խ�������ݺ�H��H�������һ�����˺���ԭ������̬���˻����������[5, 15?16]ʵ��ֵ��Ϊ�ӽ������������Hԭ���ڱ����ϵȼ�������LaNi5(111)���棬��ϵ�����������״̬���γ�ǰ��������ƽ���ȶ��ṹ����ʱHԭ�Ӽ�ľ���Ϊ0.354 0 nm���˹��̷�Ӧ������������Ӧ���������������֮�ԼΪ?2.03 eV���ڷ�Ӧ�����а�����H��H���Ķ��Ѻ�Ni��H�����γɣ������¼��ͷų������������˶ϼ�����IJ�����������������Ƿ��ȷ�Ӧ����Ӧ��ܽϵ͡�
3 ����
1) LaNi5 (111)�����ԥ�ṹLaԭ����������Niԭ����������������ƽ�ı���������˱���ԭ����Hԭ�ӵĽӴ��������������Ч�������Լ2.3%��������Hԭ�����������ɢ��
2) H2���ӽ��������Hԭ�Ӻ���LaNi5(111)�����ƽ���ȶ��ṹ���жԳ��ԣ��������⻯��LaNi5H7������ͬλ�õĽṹ��Ϊ���ƣ�Ni��Hԭ�Ӽ�������Ҫǿ��Ni��Niԭ�Ӽ������á�
3) ��ͨ��LST/ QST�����Ż��õ�����̬���о��У����������;����λ�������̬�������һ��λ�ݣ���H2������LaNi5(111)��������ԼΪ0.27 eV����ʵ��ֵ�������Ӧ������ԼΪ?2.03 eV��
REFERENCES
[1] WILLEMS J J G, BUSCHOW K H. From permanent magnets to rechargeable hydride electrodes[J]. J Less-Common Met, 1987, 129(1/2): 13?30.
[2] MALIK S K, ARLINGHAUS F J, WALLACE W E. Calculation of the spin-polarized energy-band structure of LaNi5 and GdNi5[J]. Phys Rev B, 1982, 25(10): 6488?6491.
[3] GUO J, WEI W L, MA S Y, GAO Y J. An investigation of correlation between electronic structure of LaNi4M (M=Ni, Cu, Mn, Al) and hydrogen absorption properties[J]. Mater Sci Eng B, 2003, 98(1): 21?24.
[4] Tadaei Ito, Hideaki Ido. Electronic structures and magnetic properties of LaCo5, LaNi5, and LaCo3Ni2[J]. J Appl Phys, 2005, 97(10): 10A313.1?10A313.3.
[5] SCHLAPBACH L. XPS/UPS study of the oxidation of La and LaNi5 and of the electronic structure of LaNi5[J]. Solid State Comm, 1981, 38(2): 117?123.
[6] �� ��, ������, �౾��, ������, �� ��, �Ŷ���. LaNi4.5Al0.5����Ͻ��������ܶȷ����о�[J]. �й���ɫ����ѧ��, 2007, 17(7): 1160?1165.
CHEN Dong, ZHOU Li-hai, YU Ben-hai, WANG Chun-lei, GAO Tao, ZHANG Dong-ling. Density functional theory study on solid solution phase of LaNi4.5Al0.5 hydrogen storage alloys[J]. The Chinese Journal of Nonferrous Metals, 2007, 17(7): 1160?1165.
[7] PAYNE M C, TETER M P, ALLAN D C. Iterative minimization techniques for ab initio total-energy calculations: molecular dynamics and conjugate gradients[J]. Rev Mod Phys, 1992, 64(4): 1045?1097.
[8] FISCHER S, KARPLUS M. Conjugate peak refinement: an algorithm for finding reaction paths and accurate transition states in systems with many degrees of freedom[J]. Chem Phys Lett, 1992, 194(3): 252?261.
[9] SEGALL M D, PHILIP J D, LINDAN M, PROBERT J. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Cond Matt, 2002, 14(11): 2717?2744.
[10] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Phys Rev Lett, 1996, 77(18): 3865?3868.
[11] LUE C S, SU T H, XIE B X, CHENG C. Comparative NMR study of hybridization effect and structural stability in D022-type NbAl3 and NbGa3[J]. Phys Rev B, 2006, 74(9): 094101.
[12] HAMMER B, JACOBSEN K W, NORSKOV J K. Dissociation Path for H2 on Al(110)[J]. Phys Rev Lett, 1992, 69(11): 1971?1974.
[13] HALGREN T A, LIPSCOMB W N. The synchronous-transit method for determining reaction pathways and locating molecular transition states[J]. Chem Phys Lett, 1977, 49(2): 225?232.
[14] BELL S, CRIGHTON J S. Locating transition states[J]. J Chem Phys, 1984, 80(6): 2464?2475.
[15] TANAKA S, CLEWLEY J D, FLANAGAN T B. Kinetics of hydrogen absorption by LaNi5[J]. J Phys Chem, 1977, 81(17): 1684?1688.
[16] OSOVIZKY A, BLOCH J, MINTZ M H, JACOB I. Kinetics of hydride formation in massive LaNi5 samples[J]. J Alloys and Comp, 1996, 245(1): 168?178.
������Ŀ��������Ȼ��ѧ����������Ŀ(50561002)������ʡ��Ȼ��ѧ����������Ŀ(�����0728028)��������ѧ�����ص�������Ŀ(2004ZD04)����������������������Ŀ(��̿���[2006]26-8)
�ո����ڣ�2008-01-10�������ڣ�2008-05-05
ͨѶ���ߣ��� �������ڣ���ʿ���绰��071-3232666��E-mail: guojin@gxu.edu.cn
(�༭ ������)

