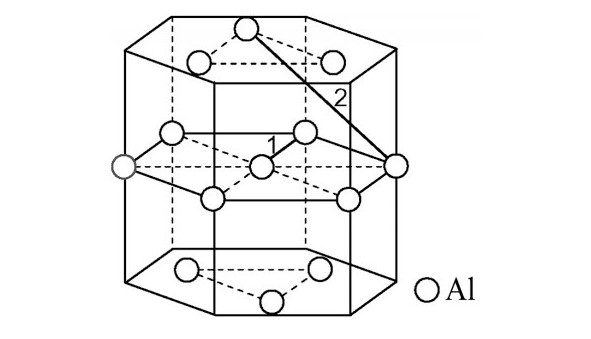
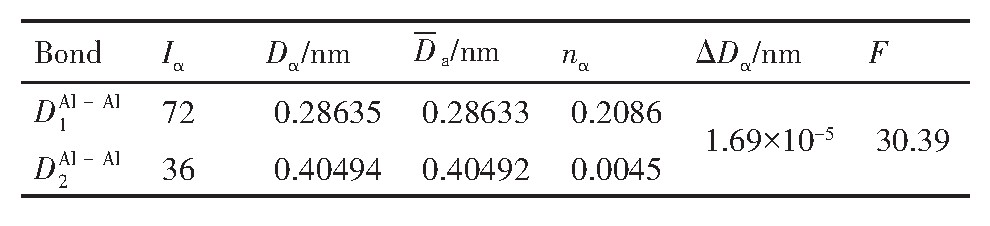
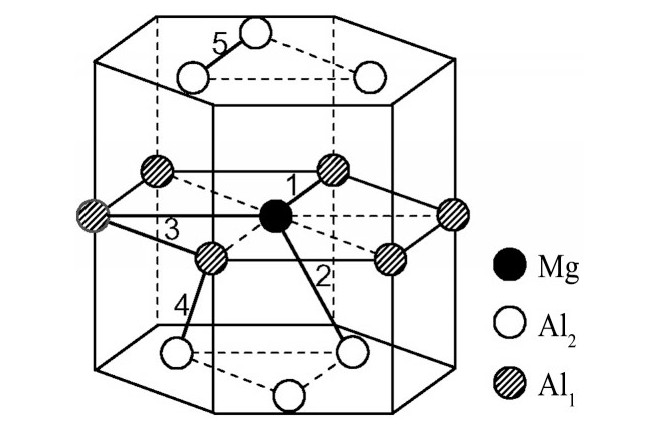
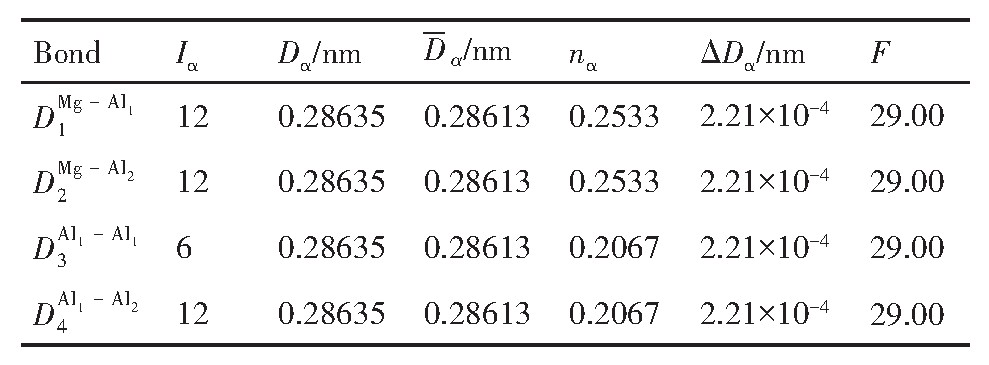
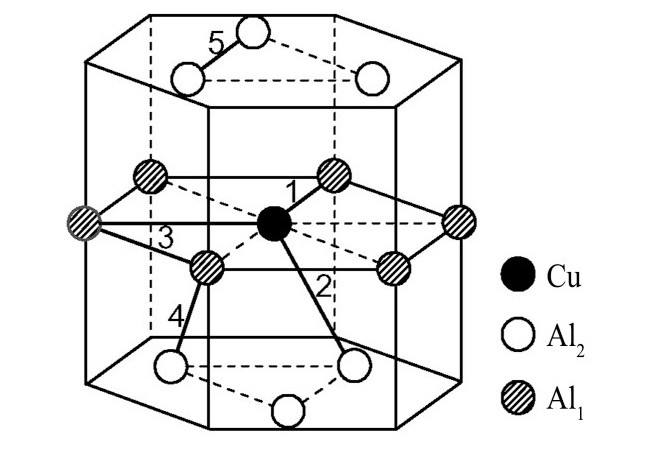
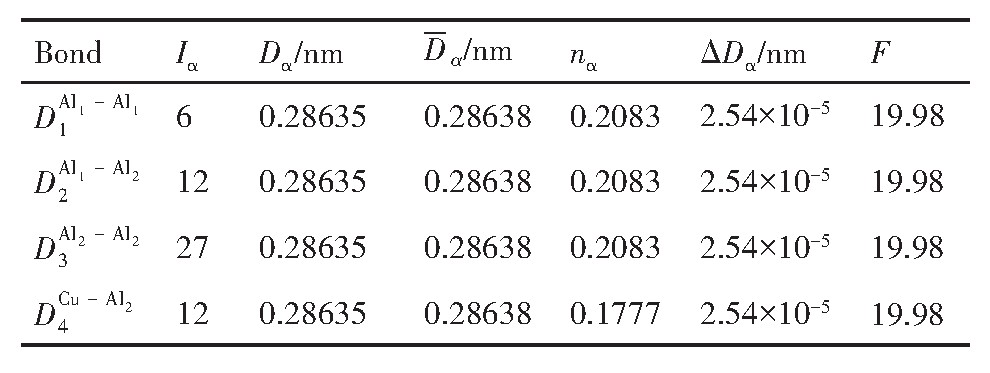
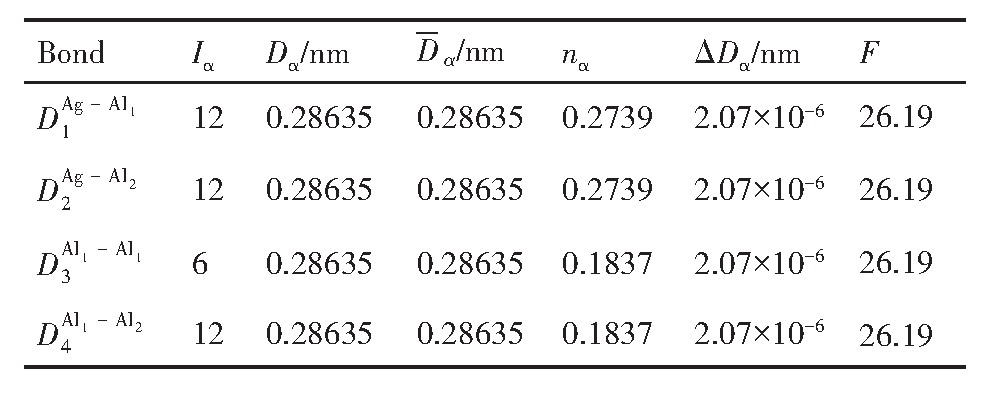
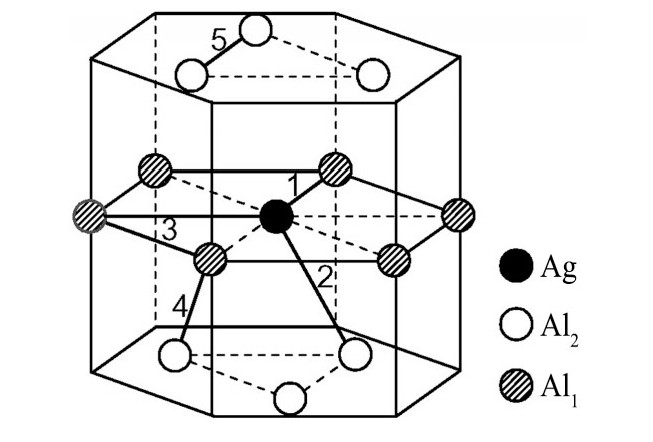
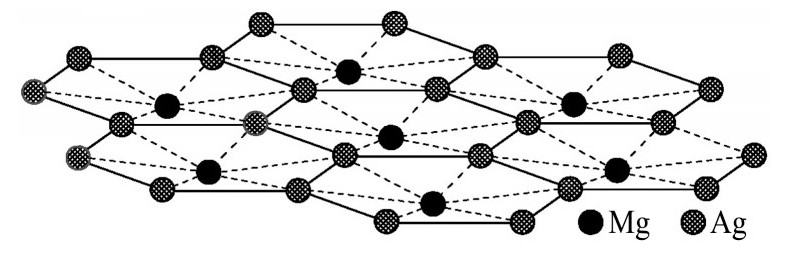
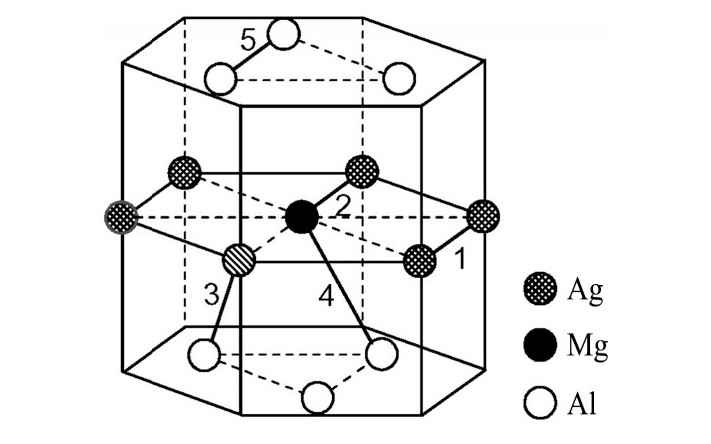
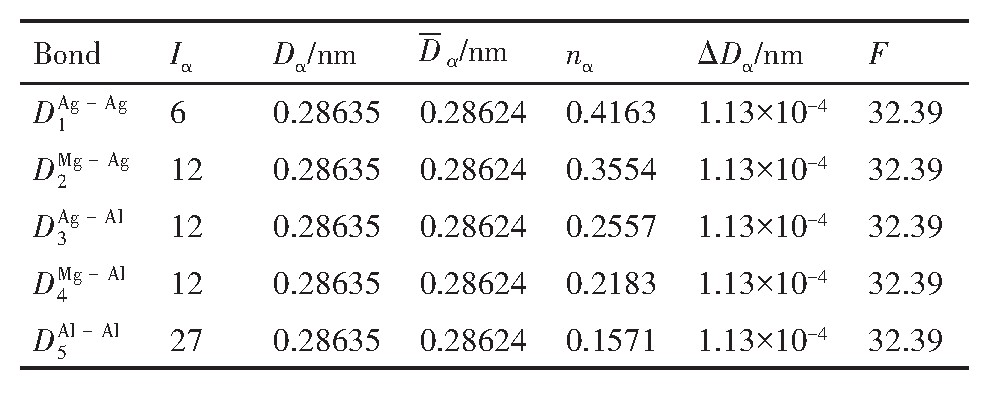
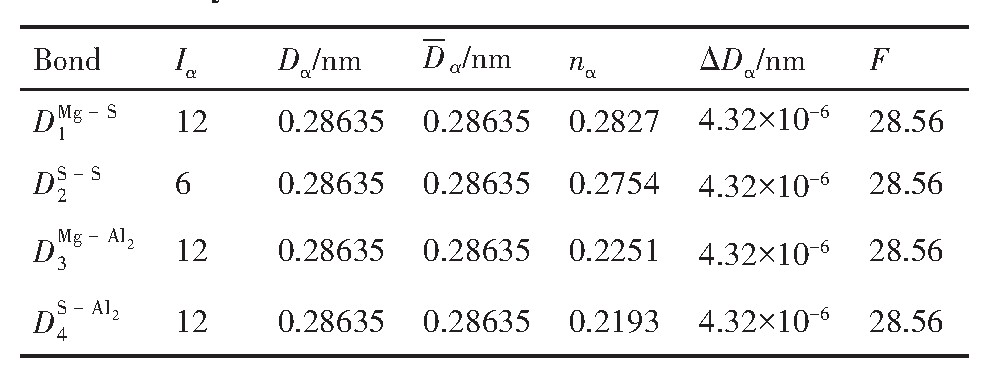
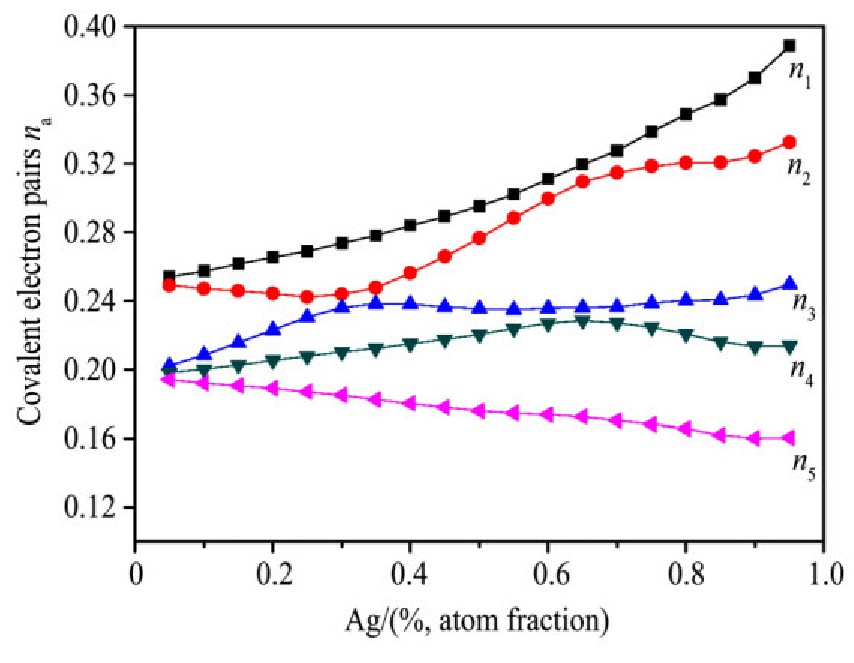
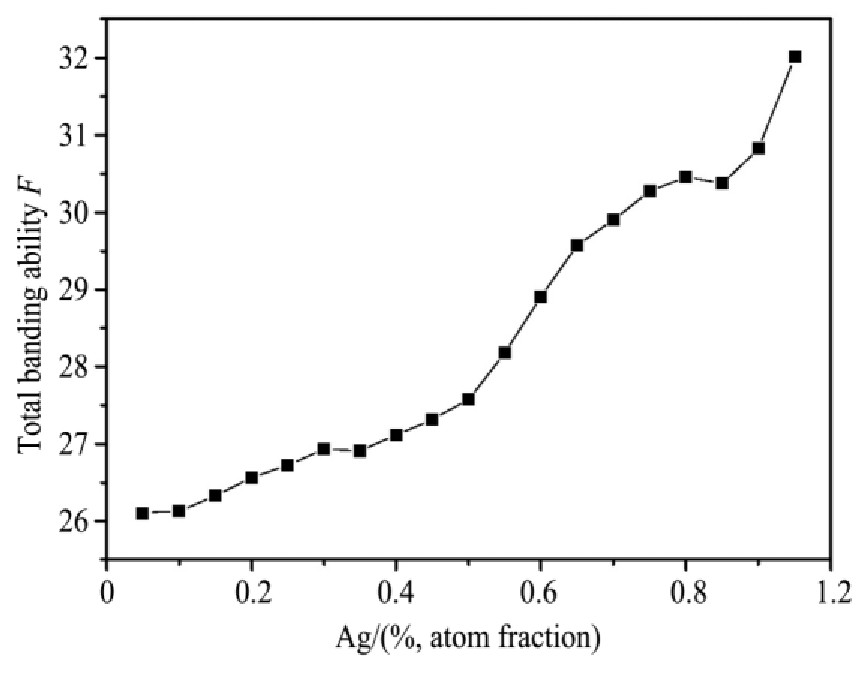
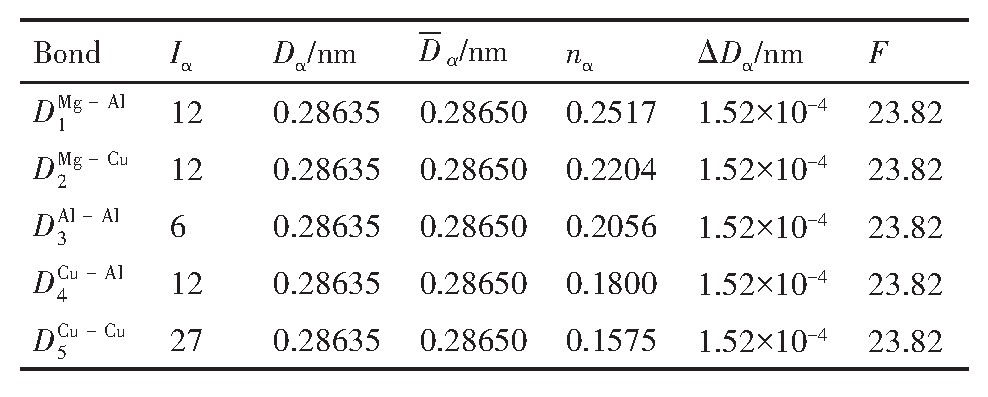
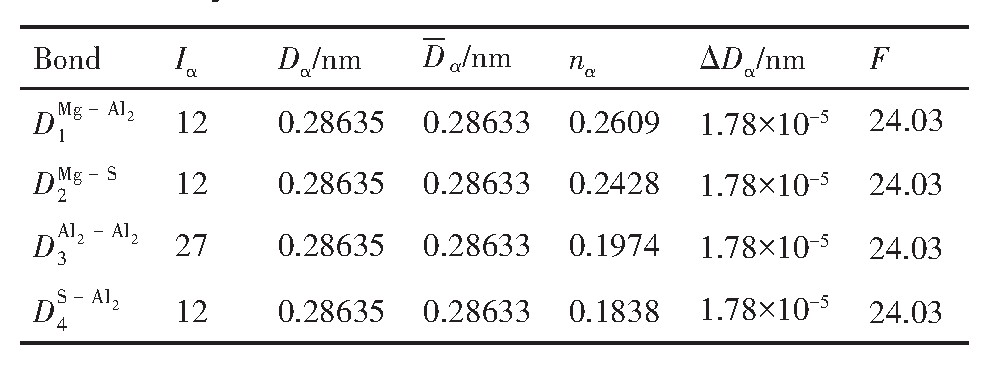
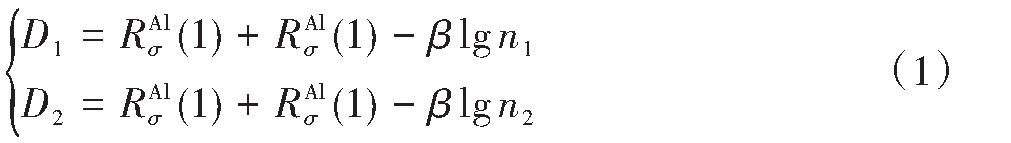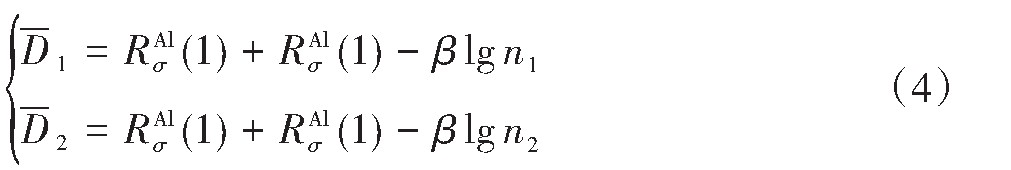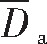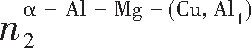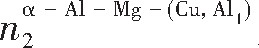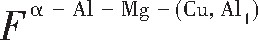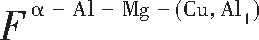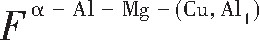ժ Ҫ������EET���ۣ�empirical electron theory of solids and molecules��,������Al-Cu-Mg-Ag�Ͻ��-Al,��-Al-Mg,��-Al-Ag,��-Al-Cu�������ʱЧ���ڦ�-Al-Mg-Ag,��-Al-Mg-Cu������ļ۵��ӽṹ,����ǿ���ۼ��Ĺ��۵�����n1�ͽṹ��Ԫ�ܳɼ�����F�����˹�����ṹ���ȶ���,�о���Ag�ڻ���{111}����ƫ�۶�Mg,Cuƫ����Ϊ�ͺϽ�ʱЧ���������Ӱ�졣�о�����:��-Al-Cu,��-Al-Ag�ͦ�-Al-Mg���ܳɼ������ֱ�Ȧ�-Al��С35.40%,15.32%��6.24%,��-Al-Cu,��-Al-Ag�ͦ�-Al-Mg�Ľṹ�ȶ��Ծ�С�ڦ�-Al�Ľṹ�ȶ��ԡ���-Al-Ag�ͦ�-Al-Mg��ǿ���ۼ���n1ֵ�ֱ�Ȧ�-Al�Ĵ�31.30%��21.43%,����-Al-Cu��ǿ���ۼ���n1ֵ�Ȧ�-Al��С0.14%,Cu��{111}�漯��û��������������ʱЧ�Ľ���,ʱЧ�����γɵĦ�-Al-Mg-Ag����ǿ������ǿ���ļ��������ܳɼ�����������,�ṹԽ��Խ�ȶ�;����-Al-Mg-Cu�����ۼ��ļ��������ܳɼ�������С,�ṹԽ��Խ���ȶ�,���ڷ��������ع���Cuԭ����Mg,Agԭ�Ӷ���{111}���ϼ���,Ϊ������γ��ṩ���κ˻���;Mg,Agԭ�Ӳ�ֱ�Ӳ��릸����κ�,��Ϊ���κ��ṩ�˴�ý����{111}����Ag,Mgԭ�ӵļ��ۼ�����{001}���ϵ�Cu��Mgԭ��,�����˦ȡ��S���������
����EET���ۣ�empirical electron theory of solids and molecules����������Al-Cu-Mg-Ag�Ͻ��-Al����-Al-Mg����-Al-Ag����-Al-Cu�������ʱЧ���ڦ�-Al-Mg-Ag����-Al-Mg-Cu������ļ۵��ӽṹ������ǿ���ۼ��Ĺ��۵�����n1�ͽṹ��Ԫ�ܳɼ�����F�����˹�����ṹ���ȶ��ԣ��о���Ag�ڻ���{111}����ƫ�۶�Mg,Cuƫ����Ϊ�ͺϽ�ʱЧ���������Ӱ�졣�о���������-Al-Cu����-Al-Ag�ͦ�-Al-Mg���ܳɼ������ֱ�Ȧ�-Al��С35.40%,15.32%��6.24%����-Al-Cu����-Al-Ag�ͦ�-Al-Mg�Ľṹ�ȶ��Ծ�С�ڦ�-Al�Ľṹ�ȶ��ԡ���-Al-Ag�ͦ�-Al-Mg��ǿ���ۼ���n1ֵ�ֱ�Ȧ�-Al�Ĵ�31.30%��21.43%������-Al-Cu��ǿ���ۼ���n1ֵ�Ȧ�-Al��С0.14%,Cu��{111}�漯��û��������������ʱЧ�Ľ��У�ʱЧ�����γɵĦ�-Al-Mg-Ag����ǿ������ǿ���ļ��������ܳɼ����������ṹԽ��Խ�ȶ�������-Al-Mg-Cu�����ۼ��ļ��������ܳɼ�������С���ṹԽ��Խ���ȶ������ڷ��������ع���Cuԭ����Mg,Agԭ�Ӷ���{111}���ϼ��ۣ�Ϊ������γ��ṩ���κ˻�����Mg,Agԭ�Ӳ�ֱ�Ӳ��릸����κˣ���Ϊ���κ��ṩ�˴�ý����{111}����Ag,Mgԭ�ӵļ��ۼ�����{001}���ϵ�Cu��Mgԭ�ӣ������˦ȡ��S���������
Valence Electron Structure and Aging Sequence of Solid Solution in Al-Cu-Mg Alloy with Addition of Ag
Qu Hua Liu Weidong Qi Jianxue Xu Qiaozhi
School of Materials Science and Engineering,Liaoning University of Technology
Abstract��
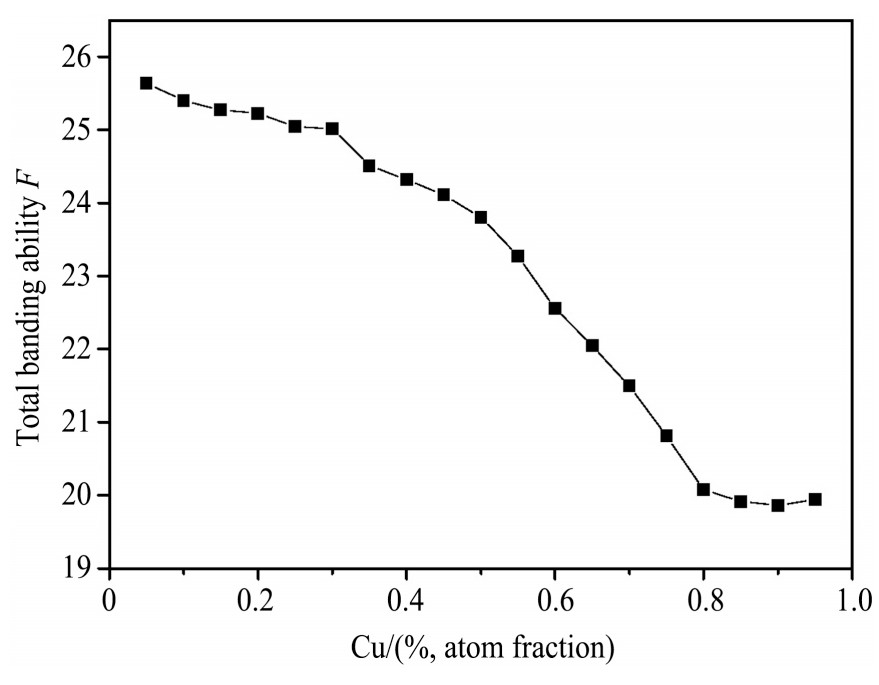
Based on the empirical electron theory of solids and molecules,the valence electron structures of solid solution of ��-Al,��-Al-Mg,��-Al-Ag,and ��-Al-Cu in Al-Cu-Mg alloy were calculated,and the valence electron structures of solid solution of ��-Al-Mg-Ag and ��-Al-Mg-Cu in Al-Cu-Mg alloy at the early aging stage were also calculated. The stability of these solid solution structures was analyzed using n1,the covalent electron number of the strongest covalent bond,and F,the total banding ability of structural unit,then the effect of Ag segregation on the{111}��plane to the segregation behavior of Mg and Cu atoms and the alloy aging sequence was studied. The results showed that the total banding abilities of ��-Al-Cu,��-Al-Ag,and ��-Al-Mg were 35.40%,15.32% and 6.24% smaller than that of ��-Al,respectively. So their structural stability was lower that of ��-Al. The n1 values of the strongest covalent bonds of ��-AlAg and ��-Al-Mg were 31.30% and 21.43% bigger and ��-Al-Cu was 0.14% smaller than that of ��-Al,respectively,therefore Cu segregation on the{111}��plane had no driving force. With the aging process,the bonding force of the strongest and the second strongest bonds and the total banding ability of Al-Cu-Mg-Ag formed in the early aging period gradually increased,and its structure became more and more stable. However,the bonding force of covalent bonds and the total banding ability of ��-Al-Mg-Cu gradually decreased,and its structure became more and more unstable,which was prone to reorganization and reconstruction. Because of Mg and Ag atoms,Cu atom gathered on{111}��plane,provided nucleation basis for the formation of �� phase. Mg and Ag atoms did not directly participate in the nucleation of �� phase,but provided �� catalyst for its nucleation. The segregation of Ag and Mg atoms on the{111}��plane reduced the Cu and Mg atoms on{001}��plane,and inhibited the precipitation of �ȡ�and S phases.
Keyword��
Al-Cu-Mg alloy;solid solution;valence electron structure;stability;aging sequence;
����EET���ۣ�empirical electron theory of solids and molecules)
[11,12]�����ӻԵ�
[13]�ӵ��ӽṹ����Ϸ����˸�Cu/Mg��Al-Cu-Mg-Ag�Ͻ��Цȡ䣬����S��ľ���������ϵ����Ӣ����
[14,15]������Ag2Al���Al-Ag�Ͻ���������Ӳ�����ü�����Χ���������γɻ��ƣ���ɵ�
[16]������Mg-Zr�Ͻ������ļ۵��ӽṹ��������Zr����ǿ���ͽ���ǿ�����ۻ��ƣ���ΰ����
[17]������Al-Cu-Mg-Ag�۵��ӽṹ�������ྺ�������Ĺ�ϵ�����ļ�����AlCu-Mg-Ag�Ͻ���������۵��ӽṹ��̽����Ag���ӶԺϽ���������۵��ӽṹ��ʱЧ���������Ӱ�졣