DOI��10.19476/j.ysxb.1004.0609.2017.08.07
��ȱ�ݶ�B2-CoSc����������Ӱ��ĵ�һ��ԭ���о�
�����ʣ���ѧ�棬�챣
(����������ѧ ���Ͽ�ѧ�빤��ѧԺ������ 454000)
ժ Ҫ�����õ�һ��ԭ����Castep��������B2�ͽ����仯����CoSc��16�ֵ�ȱ�ݵ�����ѧ���������ӽṹ�͵��Գ������м��㣬����16�ֵ�ȱ�ݴ��ڵ����ͼ��Ի�������ѧ���ܵ�Ӱ�졣���������Co����λ�γ��Ⱥͽ���ֱܷ���-6.78 eV��-0.43 eV��Co����λ�������������γɡ��ȶ�����ã������Co����λ������γ��Ⱥͽ���ֱܷ���-6.152 eV��2.504 eV���Ӷ��ó�16�ֵ�ȱ�����ȶ�������ʽ��Co��λ��Co��λ�����ڵ���̬��Co����λ��Co˫��λ��Co����λ��Co˫��λ���ɵ���̬�ܶ�ͼ�еķ����ܼ�������϶Ҳ�����жϳ���Co��λ�ͷ�λȱ�ݻ������Sc��λ�ͷ�λȱ�ݻ������ȶ�������6�ֵ�ȱ�ݵIJ��ɱ� ��֪�� Co����λ�Ļ������������ǿ��������á���������CoSc�����仯����������ȣ��п�λȱ�ݵĽ����仯�������Եõ���ߡ�
��֪�� Co����λ�Ļ������������ǿ��������á���������CoSc�����仯����������ȣ��п�λȱ�ݵĽ����仯�������Եõ���ߡ�
�ؼ��ʣ�B2-CoSc�����仯�����һ��ԭ������ȱ�ݣ�����
���±�ţ�1004-0609(2017)-08-1589-08���� ��ͼ����ţ�TG146.1���� ���ױ�־�룺A
���������������ձ����¹����й��ȶ�����B2�����仯���������ϵͳ�Ŀ������о�����ȡ�úܴ�չ[1]��Ȼ����������Ȼ���谭�����仯�����Ϊ�㷺�òĵĹؼ�����ɲ�ͬ�����仯�����������Բ��ԭ��[2]�У��۽ṹ���ӵľ��������Ա���ʱ�ܿ����Ļ�����Ŀ����[3]����λ���������������ѣ�λ�����Ľṹ�Ƿ�ƽ��ֲ������������ޣ���������ǿ�ȵͣ�����ƫ����ɾ������[4]�͵�ȱ�ݴ��ڵȡ���ˣ�����������仯���ﶼ���Խ������ģʽʧЧ������Ҳ���Ծ�����ģʽʧЧ����ʹ�Ǹ߶Գƾ���ṹ�Ľ����仯�����ڱ�����Ҳ�ȴ�����������Ͻ����Ա���������������̸�����ϣ��������ڶ�������ԶԲ��ϵ���������ҪӰ�죬������ڶ�����ִ��������ྦྷ���ȱ�ݶԲ��ϵ�Ӱ��Ҳ�ܴ�Ҳ�����������ྦྷ���ȱ�ݵȲ��ɿ����صij��֣����о����Խ����仯�������Դ��������ѣ������о������ɢ����ȷ�������ܻ�����Խ����仯����ı������ܡ�
B2�͵Ľ��������ڸ����µ��Ŵؽṹ���д�����ԭ�ӿ�λ��ͬ��Ҳ���д�����ԭ�ӷ�λ[5]����ˣ�ͨ���������̼��������Խ��ֿ�λ�ͷ�λ������������Сƫ�����Ի�����ɷ��»�õ���ྦྷ�����仯����Ӷ�ʹ�о��������Խ����仯����ı������ܳ�Ϊ���ܡ����ǣ����ֺ��д�����λ�ͷ�λ�Ľṹ��Ȼ������ܲ�����Ҫ��Ӱ�죬���ž���[6]���ڽ����仯����ṹ���Ϸ�λȱ�ݼ��������Ӱ��ı���ָ��������ȱ�ݶԽ����仯����������������ż�����Ҫ�����ã���������ȱ�ݴ��ڱ���Ϊ�ǵ��´�����Ͻ����´��Ե���Ҫԭ������������ϵ���������ѧ���ʶ����ȱ��ֱ�����[7]����ˣ��б�Ҫ��CoSc�Ͻ��ȱ�ݽṹ�Ĵ�����̬����ԺϽ������Ե�Ӱ����������о�[8]����ͨ�������Ե���ྦྷCoSc�����仯������о�����ʾ�������Բ��ϵı�������������Ȼ�������о������仯��������з��֣�����Sc����ʧ��������ԭ���ѻ�õ�һ��ȱ�ݵ�CoSc�����仯�����ˣ��������������»�õIJ������ܣ����ܴ�����ȱ�ݶԽ����仯���ﱾ�����ܵ�Ӱ�죬�Ӷ�ʹʵ�������е������³������⡣�������ԭ�����������û����ܶȷ�����һ��ԭ��ƽ�沨���Ʒ�����CoSc����ṹ��16�ֵ�һ�ĵ�ȱ��ģ�ͽ���ģ����㣬����������CoSc�����仯������жԱȣ�����16�ֵ�ȱ���ȶ�������ʽ���������Ե��ۻ��ƣ����ڻ�ö�CoSc�����仯�����ʹ���ԵĽ�һ����ʶ��
1 ����ģ���뷽��
���û����ܶȷ������۵ĵ�һ��ԭ������ƽ�沨�������Ծ��п�λ�ͷ�λȱ�ݵ�CoSc���������Ͻ�����ѧ����(�γ��ȡ��γ��ܺͽ����)�͵��ӽṹ�ļ��㡣���ӽ�����������ʹ��GGA�е�PBE�����ݶȽ�����ʽ��ģ���Ż���Brillouin�����ֲ���Monkhorst-Pack-Grid��3��3��3���зָ����������������Ⱥ���Ǣ��������е���������Ϊ2.0��10-5 eV��ƽ�沨��ȡ����EcutΪ330 eV�������������0.1 GPa�������ȡ��λ�ò�����0.01 nm��������ÿ��ԭ���ϵ���������0.005 eV/nm��ģ��ѡȡ��������CoSc�����Ŀռ��ȺΪ221��Coԭ��������(0��0��0)��Sc��ԭ��������(0.5��0.5��0.5)��B2�ṹ�ɿ��������־��������ṹ�IJ�ͬԭ��ģ��������γɵġ��ڵ�ȱ�ݼ�������У�MATTSSON��[9]����ģ�ͳߴ�Ĵ�С�����������Ӱ����Ժ��Բ��ƣ���ˣ�Ϊ�˼��㷽�㣬���о������ȱ��ʱ����2��2��2����ģ��

ͼ1 B2-CoSc��������16�ֵ�ȱ�ݽṹ����ģ��
Fig. 1 Calculation models of point defective structures of B2-CoSc
2 ��������
2.1 CoSc����ṹ�Ļ�������
�����Ż���õ�16�ֵ�ȱ�ݵ�B2-CoScƽ�⾧�����У�ֻ��Sc����λ��Sc˫��λ(��һ����)��Sc����λ�ľ��������Ż�ǰ�ľ�������ȷֱ�Ϊ0.934%��4.09%��5.06%����ʵ��ֵ(a0=0.632nm)[10]�IJ����5%���²������Ա�������CoSc��������������ѡ��������������
2.2 ��ȱ����̬�ṹ
2.2.1 ��λȱ��
�Ӳ�����λȱ�ݵ���Ϊ�����������������о�����ʽ(1)��ʽ(2)������2��2��2��������ϵCo��Sc�еĵ���λ��˫��λ������λ��ƽ����λ�γ��Ȧ�H���γ��ܦ�E[11]�����ȶ��Դ��ڽǶȷ���������ʽ(3)��������ϵ��Co��Sc����λ��˫��λ������λ��ƽ����λ�γ��ܦ���A����ʽ���£�
 (1)
(1)
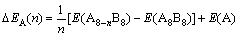 (2)
(2)
 (3)
(3)
ʽ�У���HA(n)����EA(n)�ͦ���A(n)�ֱ��ʾ����A��λ�Ļ�������γ��ȡ��γ��ܺͽ���ܣ�E(A8-nB8)��E(A8B8)�ֱ�Ϊ��λȱ�ݺ����������ﳬ����������E(A)��E(B)�ֱ��ʾA��B���������ÿ��ԭ�ӵ��������±�Ϊ�����е�ԭ�Ӹ�����n��ʾ�����п�λ������
�γ��ȡ��γ��ܺͽ���ܼ�������ͼ2����ͼ2(a)�пɼ���Co����λ��Coƽ��˫��λ��Co����λ��Sc����λ��Scƽ��˫��λ��Sc����λ������Ŀ�λ�γ��ȷֱ�Ϊ-6.78��-2.655��-1.25��-4.172��-0.089��1.057 eV����Ȼ��Co��λ�γ��ȱ�Sc��λ�γ���С�Ķ࣬����Co��λ��ȱ�ݱ�Sc��λ��ȱ�ݸ������γɡ���Co��λ�γ��Ⱥ�Sc����λ�γ��ȶ����ſ�λ�������Ӷ����ߣ�˵�����ſ�λ�������ӣ���λȱ�ݵ��γ��������͡���ͼ2(b)�ɼ���Co����λ��Coƽ��˫��λ��Co����λ��Sc����λ��Scƽ��˫��λ��Sc����λ�Ŀ�λ�γ��ֱܷ�1.87��1.08��1.64��4.48��4.23��3.94 eV���γ�Coȱ�ݻ���������Ҫ���γ������ſ�λ�������Ӷ����٣��Ҷ����γ�Sc��λȱ�ݻ������С��Ҳ����Co��λ�γ�������Sc��λ��ǿ����ͼ2(c)�ɼ���Co����λ��Coƽ��˫��λ��Co����λ��Sc����λ��Scƽ��˫��λ��Sc����λ�Ŀ�λ����ֱܷ�Ϊ-0.43��-0.37��-0.29��-0.28��-0.09�� -0.04 eV����������ó�Co��λ��������ȶ��Ա�Sc��λ�á���ˣ�ʵ����������CoSc�Ͻ��Sc�Ӿ����Ϸ���Co��λȱ��[12]�����ſ�λ�������ӣ���λ�γ������ߣ���Sc������λ�γ���Ϊ��ֵ1.0572 eV����λ��Խ���λ�ṹԽ���γɣ��γɿ�λȱ�ݻ�����ʱ��������Խ��ȶ���Խ���á��Ա�3�ֲ�ͬ��λ����֪��Co����λ�������γ����� -6.78 eV���������γɣ���Sc����λ�������γ�����1.0572 eV������ʵ�֣�Co����λ������������ -0.43 eV���ȶ�����ã������Co˫��λ������Ľ����-0.37 eV����Sc����λ������Ľ������-0.04 eV���ȶ�����
��һ���Ƚ�Co˫��λ���ݣ����ǵ�˫Co��λ��3�ֲ�ͬ����̬(Co˫��λ��һ���ڡ�Co˫��λ�ڶ����ں�Sc����λ��������)���ֱ������3����̬���γ��ȣ��γ��ܺͽ���ܣ�����������1��ͼ2�����ߵ�����ͱ�1������չʾ���γ��ȡ��γ��ܺͽ���ܵĹ�ϵ���γ��Ⱥͽ����ԽС���γ�����Խǿ���ȶ���Խ�ߡ��Աȷ�����Co-CoΪ��һ����ʱ��˫��λ��̬�γ��Ⱥͽ���ֱܷ�Ϊ-0.14��-0.39 eV����ṹ�γ�������ǿ���ȶ�����á���Co-CoΪ��������ʱ�Ļ������γ��Ⱥͽ����Ϊ0.127��-0.36 eV���γ��������ȶ�����
��1 Sc˫��λ��Co˫��λ���γ��ȣ��γ��ܺͽ����
Table 1 Formation heat, formation energy and binding energy of Sc with double vacancies and Co with double anti-sites(eV)
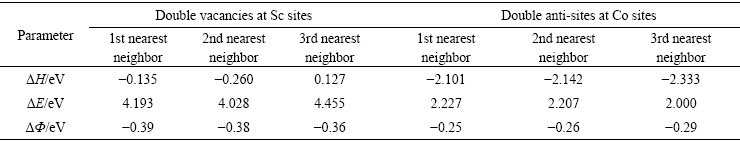
ͼ2 Co8-nSc8��Co8Sc8-n��������λ��˫��λ������λ�Ŀ�λ����
Fig. 2 Energy per vacancy in Co8-nSc8 or Co8Sc8-n(n=1, 2, 3) supercells with n vacancies
2.2.2 ��λȱ��
ͬ�����Ӳ�����λ���������Ƕȷ������������߲���ʽ(4)��ʽ(5)�ֱ������2��2��2������Co��Sc��ϵ�������еĵ���λ��˫��λ������λ��ƽ����λ�γ��Ȧ�H���γ��ܦ�E[11]�����ȶ��Դ��ڽǶȷ������������߲���ʽ(6)�ֱ�����˳�������ϵ��Co��Sc����λ��˫��λ������λ�Ľ���ܦ�������ʽ���£�
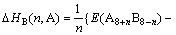
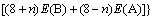 (4)
(4)
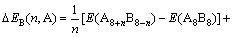
 (5)
(5)
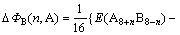 (6)
(6)
ʽ�У���HB(n, A)����HB(n, A)�ͦ���B(n, A)�ֱ��ʾn��Aԭ��ռBλ��ƽ����λ�γ��ȡ��γ��ܺͽ���ܡ�E(A8B8)��ʾ8��A(Co)��8��B(Sc)ԭ����ɳ�������������������ͼ3��ʾ��
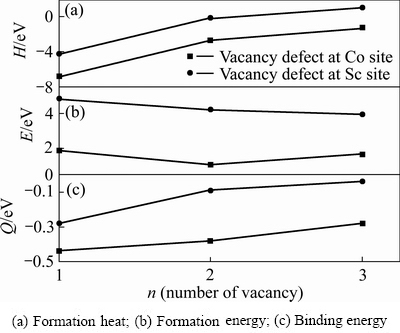
��ͼ3��Co��λ��Sc��λ��ƽ����λ�γ��ȡ��γ��ܺͽ���ܿ��Կ����������ǵ���λ��˫��λ��������λ������ȱ�ݣ�����Co��λ����Ҫ���������Ȳ���Sc��λ����Ҫ�����ͣ�����Co��λ�����γɣ�ȱ���ȶ��Ժá�ͼ3�У�����Co��λȱ���������࣬ȱ���γ��ȡ��γ��ܺͽ���������ߣ��γ��������ȶ������ͣ���ζ�Ÿ�Ũ�ȵķ�λȱ�����շ�����롣��ͼ3(a)��3(b)�ɼ����γ�Co����λ��������Ҫ���γ��Ⱥ��γ��ֱܷ�Ϊ-6.152 eV��1.504 eV������������γɣ����ȶ�����á���ͼ3(c)֪��Co����λ���������ȶ�ʱ��Ҫ�Ľ����Ϊ-0.37 eV���ڷ�λȱ����������������͵ģ�����Co����λ�������ȶ�����ã����γ�Sc����λȱ�ݻ�����ʱ��Ҫ�Ľ����Ϊ-0.16 eV���ȶ�����
���Ƶ����Co˫��λ��3�ֲ�ͬ��̬(Co˫��λ��һ���ڡ�Co˫��λ�ڶ����ں�Co����λ��������)������3����̬���γ��ȡ��γ��ܺͽ���ܣ�ֵ���ڱ�2���ɱ�2�ɼ���Co-CoΪ�����ڽ�ʱ˫��λ�γ��ȡ��γ��ܺͽ������ͣ��ֱ�Ϊ-2.333 eV��2.000 eV��1.995 eV��������3����̬��Co-Co˫��λ����������̬�������γɲ����ȶ�����ã����ΪCo-Co˫��λ�ڶ�������̬��������ΪCo-Co˫��λ��һ���ڡ��Ա������������ݿ�֪����λ��̬���γ��Ⱥ��ȶ�����õ���˫Co-Co��λ�ĵ�������ȱ�ݣ��γ�����ǿ����Co����λ���ȶ���������Sc����λ��
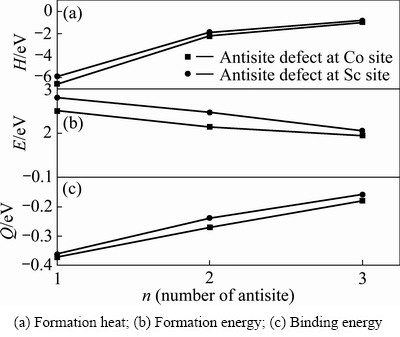
ͼ3 Co8-nSc8+n��Co8+nSc8-n��������λ��˫��λ������λ������
Fig. 3 Energy per anti-site defect in Co8-nSc8+n or Co8+nSc8-n(n=1, 2, 3) supercells with n anti-site defects
��ͼ2��3�п�λ�ͷ�λ���γ��ܺͽ���ܿɼ���ƫ��CoSc���뻯ѧ�����Ϊ1:1�ĸ���Co�Ͻ���ܳ��ֵ�ȱ������ΪCo��λ��Sc��λ[13]�������о���8�ַ�λ��8�ֿ�λ�е�5��Co��λ��5��Sc��λ�γ��ܣ���Co����λ��Co˫��λ�������ڡ�Co˫��λ�ڶ����ڡ�Co˫��λ��һ���ڡ�Co����λ��Sc����λ��Sc˫��λ�ڶ����ڡ�Sc˫��λ��Sc˫��λ�������ں�Sc����λ���γ��ֱܷ�Ϊ1.94��1.99��2.21��2.23��2.50��3.94��4.03��4.19��4.46��4.48 eV���Աȿ�֪Co��λ�γ��ܱ�Sc��λ�γ��ܵͣ�Ԥ�⸻��Co�Ͻ��CoSc�Ͻ��п��ܳ��ֵ�ȱ������ΪCo��λ��ͬ����ƫ��CoSc���뻯ѧ����ȵĸ���Sc�Ͻ���ֵ�ȱ������ΪSc��λ��Co��λ��8�ַ�λ��8�ֿ�λ�е�3��Sc��λ��3��Co��λ�γ��ܣ���Sc����λ��Sc˫��λ��һ���ڡ�Sc����λ��Co˫��λ��Co����λ��Co����λ�Ľ���ܣ�����ֵ�ֱ�Ϊ2.05��2.47��2.81��1.48��1.64��1.88 eV���Ա�6�����ݿ�֪��Co��λ����ܱ�Sc��λ����ܵ���30%���ң�Ԥ�⸻��Sc�Ͻ��CoSc�Ͻ��г��ֵ�ȱ������ΪCo��λ���Ӷ��Ʋ��ƫ���������Ȼ������ȱ���������п��ܵ���Co��λ��Co��λ�����ڵ���̬��Co����λ��Co˫��λ��Co����λ��Co˫��λ��
2.2.3 ȱ�ݵĵ��ӽṹ
Ϊ�˴ӵ��ӽṹ��ʾ��ͬ��ȱ���ȶ��Դ�����ʽ���о��˿�λ�ͷ�λȱ�ݵ�̬�ܶȷֲ�(DOS)��ͼ4��ʾΪ�����仯����CoSc�д���Co��λ��Co��λ��Sc��λ��Sc��λȱ����ϵ����̬�ܶȼ�������ͼ4���鴹ֱ�������ߵĽ����Ӧ�����������ֵ�Ƿ�����N���ɴ˿��Կ�����4��Co7Sc8��Co9Sc7��Co8Sc7��Co7Sc9̬�ܶ�ͼ�ķ���ʮ��������ҷ����ܼ�fλ�ڵ��ܳɼ�̬���ܷ���̬����ijɼ��崦[14]��ͼ4(a)��ʾΪ����Scȱ�ݺϽ��̬�ܶ�ͼ���Ա�Co��λ��Sc��λ��̬�ܶ����߹�ϵ��Co��λ���ķ�������ֵ��Sc��λ�ڷ�������ֵƫ�ͣ�˵��Co��λ�Ͻ��Sc��λ�Ͻ��ȶ���ͼ4(b)��ʾΪ����Coȱ�ݺϽ��̬�ܶ�ͼ���Ա�Co��λ��Sc��λ��̬�ܶ����߹�ϵ��Co��λ���ķ�������ֵ��Sc��λ�ڷ�������ֵ�ͣ�˵��Co��λ�Ͻ���ȶ��Ա�Sc��λ�Ͻ���ȶ��Ժá�������ȱ�ݽṹ�и������ҵ�Co�Ŀ�λ�ͷ�λȱ����ʽ�����ӽṹͼ���������ǰ����������������Ǻϡ����⣬����϶��ֱ�ӷ�Ӧ��ϵ���ۼ�ǿ��������϶Խ�����ۼ�Խǿ[15]����̬�ܶ�ͼ�����ó�4�ֻ�����Co7Sc8��Co9Sc7��Co7Sc9��Co8Sc7������϶�ֱ�Ϊ2.0��1.9��1.7��0.7 eV�����ۼ���ǿ����ΪCo7Sc8��Co9Sc7��Co7Sc9��Co8Sc7�����ۼ�Խǿ����ϵԽ�ȶ�����һ����֤���ɽ���ܺ͵���̬�ܶ����ó����ȶ��Խ��ۡ�
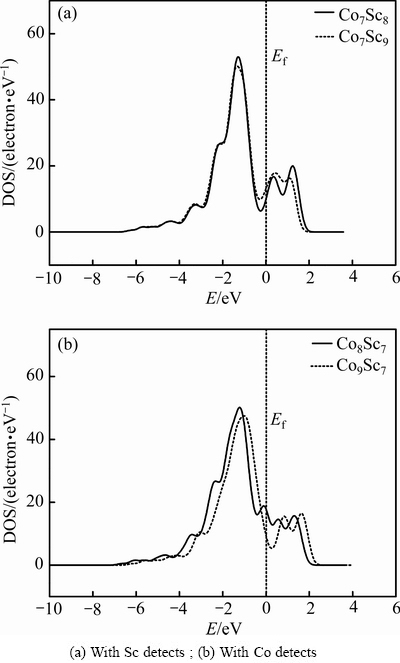
ͼ4 Co7Sc8��Co9Sc7��Co8Sc7��Co7Sc9�����仯�������̬�ܶ�ͼ
Fig. 4 Total state densities of Co7Sc8, Co9Sc7, Co8Sc7 and Co7Sc9 intermetallic compounds
3 ȱ�ݽṹ�ı�����������
����B2�ṹ�Ľ����仯���������������ṹȱ�ݵĿ�λ��λԭ�����ͽ������[16]��GSCHNEIDNER��[17]���֣����гɷ�ȷ������ȫ�����B2�ṹRM(RΪϡ��������MΪ��͢�~����Ľ���)�Ľ����仯���������¾��зdz��õ������ԡ����ڲ��ɱ�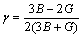 ���о������仯����������������ijɹ�[18]�����ڿ�λȱ�ݺͷ�λȱ�ݴ�����ʽ�����о�������Co����λ��Co˫��λ��Co����λ��Co˫��λ(��һ���ڡ��ڶ����ں͵�������)��6�ֵ�ȱ�ݵIJ��ɱ�
���о������仯����������������ijɹ�[18]�����ڿ�λȱ�ݺͷ�λȱ�ݴ�����ʽ�����о�������Co����λ��Co˫��λ��Co����λ��Co˫��λ(��һ���ڡ��ڶ����ں͵�������)��6�ֵ�ȱ�ݵIJ��ɱ�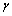 ������������2������Co˫��λ��һ���ڡ�Co����λ��Co˫��λ��Co˫��λ�������ڡ�Co˫��λ�ڶ����ں�Co����λ����ֵ�ֱ�Ϊ0.30��0.31��0.31��0.31��0.34��0.35�����������ӣ����ݽ�������ѹ��Ϊ���͵���ֵ��������ֵԽ�����������Խǿ�����ϵ���չ��Խ�ã���Co����λ�Ļ�����Ľ�������ǿ����չ����ã�����ѹ��ֵΪ��ʱ����ֵԽ���ʾ���ϵֿ���������Խ���ұ��ֳ����з����ԵĹ��ۼ������ԣ�����������CoSc�������ֵΪ-0.85������������������6�ֵ�ȱ�ݻ������еֿ������������˵����ȱ�ݵĴ��ڿ��շ�������Ĵ��Խ��ͣ�ʹ��������������ӡ����п�λȱ�ݡ���λȱ�ݺͲ��ɱȷ����õ�������Ե���Ҫ��Co��λȱ�ݶ���Scȱ��(��λ�ͷ�λ)������Pugh�����Բ���/���Բ����оݣ�6�ֵ�ȱ�ݵ�B/G������1.75�����2��ʾ��ȱ�ݻ�����������Ե����ԣ��������ɴ�С����ΪCo����λ��Co˫��λ�ڶ����ڡ�Co˫��λ�������ڡ�Co˫��λ��Co����λ��Co˫��λ��һ���ڣ������ò��ɱ��õ��Ľ����Ǻϡ�
������������2������Co˫��λ��һ���ڡ�Co����λ��Co˫��λ��Co˫��λ�������ڡ�Co˫��λ�ڶ����ں�Co����λ����ֵ�ֱ�Ϊ0.30��0.31��0.31��0.31��0.34��0.35�����������ӣ����ݽ�������ѹ��Ϊ���͵���ֵ��������ֵԽ�����������Խǿ�����ϵ���չ��Խ�ã���Co����λ�Ļ�����Ľ�������ǿ����չ����ã�����ѹ��ֵΪ��ʱ����ֵԽ���ʾ���ϵֿ���������Խ���ұ��ֳ����з����ԵĹ��ۼ������ԣ�����������CoSc�������ֵΪ-0.85������������������6�ֵ�ȱ�ݻ������еֿ������������˵����ȱ�ݵĴ��ڿ��շ�������Ĵ��Խ��ͣ�ʹ��������������ӡ����п�λȱ�ݡ���λȱ�ݺͲ��ɱȷ����õ�������Ե���Ҫ��Co��λȱ�ݶ���Scȱ��(��λ�ͷ�λ)������Pugh�����Բ���/���Բ����оݣ�6�ֵ�ȱ�ݵ�B/G������1.75�����2��ʾ��ȱ�ݻ�����������Ե����ԣ��������ɴ�С����ΪCo����λ��Co˫��λ�ڶ����ڡ�Co˫��λ�������ڡ�Co˫��λ��Co����λ��Co˫��λ��һ���ڣ������ò��ɱ��õ��Ľ����Ǻϡ�
��2 6�ֵ�ȱ�ݽ����仯�����ģ�����ļ�����
Table 2 Calculation results of modulus and of six kinds of intermetallics with point defects

4 ����
1) �ɿ�λ�γ��Ⱥ��γ������ݿ�֪����CoSc�����仯�����Ӹ����������γ�Co����λ�����ӽ����Ԥ�⣬Co˫��λ�ȶ�����á��ɷ�λ�γ��Ⱥͷ�λ��������ݷ�����֪������Co��λ����Ҫ������������Sc��λ����Ҫ������һ������ˣ���CoSc�����仯���������γ�Co��λ���ͷ�λԭ�Ӷ��ԣ�Co����λ���ȶ���ȱ����ϵ����̬�ܶ�ͼ�ͷ����ܷ���Ҳ֤ʵ������ѧ��������ȷ�ԡ�
2) ����ƫ��CoSc���뻯ѧ����ȵ��Ǹ���Co���Ǹ���Sc�ĺϽ𣬽����仯����CoScȱ�����Ͷ�����Co��λ��Co��λȱ����ʽ���ڣ�����̬��Co����λ��Co˫��λ��Co����λ���Լ�Co˫��λ��
3) ��������CoSc�����仯�����ֵΪ-0.85��Ƚϣ�Co˫��λ��һ���ڡ�Co����λ��Co˫��λ��Co˫��λ�������ڡ�Co˫��λ�ڶ����ں�Co����λ��6�ֵ�ȱ��CoSc������IJ��ɱ�ֵ��Ϊ��ֵ�������бȽ�ǿ�Ľ������ɷ֣��ó���ȱ�ݵĴ��ڿ����������������ԣ�����������������Ҫ��Co��λȱ�ݡ���Push������֪��ȱ��CoSc������B/G����1.75�����ֳ����Ե����ԡ�
REFERENCES
[1] �� ��, Ҧ����, ��ƽ��, κ����, ����ϣ, �����. Cr��Mo��W��FeAl�����仯������ӽṹ����ѧ����Ӱ��ĵ�һ��ԭ���о�[J]. ϡ�н������Ϲ���, 2014, 43(9): 2112-2117.
CHEN Yu, YAO Zheng-jun, ZHANG Ping-ze, WEI Dong-bo, LUO Xi-xi, HAN Pei-de. First-Principles study on effects of Cr, Mo and W on the electronic structure and mechanical properties of FeAl intermetallic compounds[J]. Rare Metal Materials and Engineering, 2014, 43(9): 2112-2117.
[2] ��ʺ�, �� ��, �� ��. B2��TiSi�Ͻ��ȱ�ݽṹ����ѧ���ܵĵ�һ��ԭ���о�[J]. ����ɽ��ѧѧ��(��Ȼ��ѧ��), 2014, 35(6): 77-80.
SUN Cai-hong, WU Min, AN Bo. First-principle study on the point defective structures and mechanical property of B2-TiSi alloy[J]. Journal of Jinggangshan University (Natural Science), 2014, 35(6): 77-80.
[3] ֣����, �� ƽ, �� ΰ, �����, ���¿�, ��С��, ������. Al������CrAlNͿ���۽ṹ����ѧ���ܵ�Ӱ��[J]. ��տ�ѧ�뼼��ѧ��, 2011, 31(6): 686-689.
ZHENG Kang-pei, LIU Ping, LI Wei , MA Feng-cang, LIU Xin-kuan, CHEN Xiao-hong, YANG Li-hong. Impacts of Al content on microstructures and mechanical properties of CrAlN coatings[J]. Chinese Journal of Vacuum Science and Technology, 2011, 31(6): 686-689.
[4] ����ͤ. ������Ԫ���ڸ��ºϽ��е����������[J]. �й���ɫ����ѧ��, 2011, 21(3): 465-475.
GUO Jian-ting. Effects of several minor elements on superalloys and their mechanism[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(3): 465-475.
[5] ��˳ƽ, ��Сƽ, �� �S, ¬����, � ��, ����, �� ��. L12-Al3Li�����仯�����ȱ��Ũ�ȵĵ�һԭ������[J]. �й���ɫ����ѧ��, 2013, 23(2): 370-378.
SUN Shun-ping, LI Xiao-ping, YU Yun, LU Ya-lin, ZANG Bing, YI Dan-qing, JIANG Yong. First-principle calculation of point defects concentration in L12-Al3Li intermetallic[J]. The Chinese Journal of Nonferrous Metals, 2013, 23(2): 370-378.
[6] �� ��, �� �, �� ��. �����仯����ṹ���Ϸ�λȱ�ݼ�������ܵ�Ӱ��[J]. ϡ�н������Ϲ���, 2013, 42(2): 429-434.
ZHANG Jing, CHEN Zheng, YANG Tao. Antisite defect in the intermetallic structural materials and its effect on the mechanical performance[J]. Rare Metal Materials and Engineering, 2013, 42(2): 429-434.
[7] �� ��, �� ƽ, ���. B2-RuAl��ȱ�ݽṹ�ĵ�һԭ������[J]. ϡ�н��������빤��, 2006, 35(7): 1066-1070.
CHEN  , PENG Ping, LI Gui-fa. First-principle calculation of point defective structures of B2-RuAl intermetallic compound[J]. Rare Metal Materials and Engineering, 2006, 35(7): 1066-1070.
, PENG Ping, LI Gui-fa. First-principle calculation of point defective structures of B2-RuAl intermetallic compound[J]. Rare Metal Materials and Engineering, 2006, 35(7): 1066-1070.
[8] KRESSE G, FURTHMULLER J. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set[J]. Computational Materials Science, 1996, 6(1): 15-50.
[9] MATTSSON T R, MATTSSON A E. Calculating the vacancy formation energy in metals: Pt, Pd, and Mo[J]. Physical Review B Condensed Matter, 2002, 66(21): 214110.
[10] GSCHNEIDNER K A , IKEDA K, TSANG T W E, McMASTRS O D, STIERMAN R J, EUCKER S S, LAMBERT S E, MAPLE M B, BUCHAL C. Influence of high magnetic fields (10 T) on paramagnons in rare-earth intermetallic compounds[J]. Physica B Condensed Matter, 1985, 130(1/3): 202-206.
[11] KELLOU A, FERAOUN H I, GROSDIDIER T, CODDET C, AOURAG H. Energetics and electronic properties of vacancies, anti-sites, and atomic defects(B, C, and N)in B2-FeAl alloys[J]. Acta Materialia, 2004, 52(11): 3263-3271.
[12] �� ��, �� ��, ������, �� ��. ��һ��ԭ���о���λ��ȱ�ݶԸ�ѹ��LiF�ĵ��ӽṹ��ѧ���ʵ�Ӱ��[J]. ����ѧ��, 2011, 60(2): 541-545.
HE Xu, HE Lin, TANG Ming-jie, XU Ming. Effects of the vacancy point-defect on electronic structure and optical properties of LiF under high pressure: A first principle investigation[J]. Acta Physica Sinica, 2011, 60(2): 541-545.
[13] �ų���, �� ��, ���Ƿ�, �ջԽ�, ��˳ƽ, Ҧ����. ��ȱ��Ũ�ȶԷǻ�ѧ������L1_2�ͽṹ��A1_3Sc�������ܵ�Ӱ ��[J]. ����ѧ��, 2016, 65(7): 238-249.
ZHANG Chao-min, JIANG Yong, YIN Deng-feng, TAO Hui-jin, SUN Shun-ping, YAO Jian-gang. Effects of point defect concentrations on elastic properties of off-stoichiometric L1_2-type A1_3Sc[J]. Acta Physica Sinica, 2016, 65(7): 238-249.
[14] ¬����, ���, �� �, �� ��. ��ȱ�ݶ�B2-NiAl��ѧ����Ӱ��ĵ�һ��ԭ������[J]. �й���ѧ: ����ѧ��ѧ����ѧ, 2013, 43(2): 152-158.
LU Yan-li, HOU Hua-xin, CHEN Zheng, MU Hong. First-principles calculation on mechanical and thermal properties of B2-NiAl with point defects[J]. Scientia Sinica Physica: Mechanica and Astronomica, 2013, 43(2): 152-158.
[15] KELLOU A, FERAOUN H I, GROSDIDIER T, CODDET C, AOURAG H. Energetics and electronic properties of vacancies, anti-sites, and atomic defects(B, C, and N)in B2-FeAI alloys[J]. Acta Mater, 2004, 52(11): 3263-3271.
[16] BAKER I. A review of the mechanical properties of B2 compounds[J]. Materials Science and Engineering A, 1995, 192: 1-13.
[17] GSCHNEIDNER K, RUSSELL A, PECHARSKY A, MORRIS J, ZHANG Z H, LOGRASSO T, HSU D, CHESTER LO C H, YE Y Y, SLAGER A, KESSE D. A family of ductile intermetallic compounds[J]. Nature Materials, 2003, 2(9): 587-591.
[18] RUSSELL A M, ZHANG Z, LOGTASSO T A, LO C C H, PECHARSKY A O, MORRIS J R, YE Y, GSCHNEIDER K A, SLAGER A J. Mechanical properties of single crystal YAG[J]. Acta Materialia, 2004, 52(13): 4033-4040.
Effect of point defects on physical and mechanical properties of B2-CoSc intermetallic studied by first-principles method
ZHU Pan-pan, GUO Xue-feng, CUI Hong-bao
(School of Materials Science and Engineering, Henan Polytechnic University, Jiaozuo 454000, china)
Abstract: First-principles based on the density functional theory was conducted to systematacially investigate the thermodynamic parameters, the electronic structures and the elastic properties of B2-CoSc intermetallic with 16 different point defects. Structural stability and elastic-plastic deformation mechanism were studied based on the calculation. The results show that the lowest formation heat and formation energy of single vacancy at Co site are -6.78 eV and -0.43 eV, respectively, so the B2-CoSc intermetallic with single vacancy at Co site is the easiest to form at stead state at room temperature condition. The formation heat and the formation energy of anti-site defect at Co site are -6.152 eV and 2.504 eV, respectively, and the intermetallic with anti-site defect at Co site is also easier to form and more stable. It is concluded that the most possible forms of point defects are vacancy and anti-site defects at Co site. Specifically, they are single vacancy, double vacancies, three vacancies and double anti-site defects at Co site. The vacancy and anti-site defects at Co site are more stable than vacancy and anti-site defects at Sc site, followed by fermi level and pseudogap of electronic state density figure. Therefore, three vacancies defect at Co site of CoSc intermetallic has the strongest metallic bonding and the best plasticity among the intermetallics with six kinds of point defects by comparing the values of poisson . So, the plasticity of intermetallics with vacancy defectes is improved, comparing with the plasticity of perfect CoSc intermetallic.
Key words: B2-CoSc intermetallic; first-principles; point defect; plasticity
Foundation item: Projects(51271073, 51301063) supported by the National Natural Science Foundation of China
Received date: 2016-05-17; Accepted date: 2016-10-07
Corresponding author: GUO Xue-feng; Tel: +86-391-3986906; E-mail: guoxuef@hpu.edu.cn
(�༭ ��ѧ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(51271073��51301063)
�ո����ڣ�2016-05-17�������ڣ�2016-10-07
ͨ�����ߣ���ѧ�棬���ڣ���ʿ���绰��0391-3986906��E-mail: guoxuef@hpu.edu.cn

