���±�ţ�1004-0609(2008)12-2224-09
����Mg�����ȶ��Եĵ�һԭ�����ӽṹ����
�ջԽ�1, 2���� ��3����־��1���Ŵ���2���� ��2���Ʋ���3
(1. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ����ɳ 410083��
2. ���ϴ�ѧ ұ���ѧ�빤��ѧԺ����ɳ 410083��
3. ���ϴ�ѧ ��ĩұ������ص�ʵ���ң���ɳ 410083)
ժ Ҫ��������С���˷���SGTE���������ݿ��н���Mg��Gibbs�ܱ���ʽ�����������������õ��˱�SGTE���ݿ����ȷ�Ľ����ͬʱ����SGTE���ݿ�ľ����ȶ�����������0 K�����һԭ����������ƽ�沨�����Ľ�������˶Աȣ����ֵ�һԭ���ľ����ȶ��������Ϊ?Gbcc?hcp��?Gfcc?hcp��0����SGTE���ƽ��һ�¡�ͬʱ���о������֣���һԭ������ƽ�沨���Ʒ��������hcp-��fcc-��bcc-Mg�ľ�������ԭ�������ʵ��ֵ�Լ�ͶӰ�Ӳ������õ��Ľ��ƫ����ܳ�������ȫ�෴�Ľ��, ����3�ֽṹ�IJ���s̬����ת��Ϊp̬�����γ��˸�ǿ�Ļ�ѧ����
�ؼ��ʣ�Mg��Gibbs�ܣ����ӽṹ�������ȶ��ԣ���һԭ��
��ͼ����ţ�TG 111 ���ױ�ʶ�룺 A
Calculations of lattice stabilities of elemental Mg from electronic structures in first principles
TAO Hui-jin1, 2, YIN Jian3, YIN Zhi-min1, ZHANG Chuang-fu2, LI Jie2, HUANG Bo-yun3
(1 School of Materials Science and Engineering, Changsha 410083, China;
2. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China;
3. State Key Laboratory of Powder Metallurgy, Central South University, Changsha 410083, China)
Abstract: The correction of transition data and reassessment of the parameters of Gibbs energy of elemental Mg were performed with the least-square method and the results agree more accurately with JANAF data than those of SGTE database. At the same time, the lattice stability parameters obtained by CALPHAD method in SGTE database were extrapolated to 0 K, and these results were compared with those of total energy plane wave pseudopotential method in first principles. It is found that the result of first principles agrees completely with that of SGTE, ?Gbcc?hcp��?Gfcc?hcp��0. Besides, it is found that the results of lattice constants and atomic volumes of hcp-, fcc- and bcc-Mg calculated by total energy plane wave pseudopotential method are much larger than experimental data and those of projector augmented wave method in first principles. The contrary case occurs in total energy. And part of s state electrons in atoms were changed into p state electrons in crystals to form stronger chemical bonding.
Key words: Mg; Gibbs energy; electronic structure; lattice stability; first principles
��ͼ�ļ���ģ��(CALPHAD��CALculations of PHAse Diagrams)һֱ����Ϊ��ָ���²��Ͽ�����Ƶ�ǿ��������[1?2]���Դ����ʲ�ͬ����ṹ���Gibbs�ܣ��������ȶ�������������CALPHAD����Ҫ���������У�SGTE(Scientific Group Thermodata Europe) ���������ݿ�[3]�Ѿ�����298.15 K����78��Ԫ�صľ����ȶ������Ͳ�ͬ����ṹ��Gibbs�ܱ���ʽ���뽨���ڶ�ʵ��������ϲ��������ƻ����ϵ�CALPHAD������Ϊ������ǣ���һԭ�������ڲ�����ʵ�����ϵ�����¿��Խ��д����ʲ�ͬ����ṹ�����ܡ���������̬�ܶȵ��������ʵ����ۼ��㣬���õ����ⴿ���ʾ����ȶ��Ե�����ԭ��[4?13]��
����SGTE���ݿ������ڽ���Mg Gibbs��������JANAF (Joint Army Navy Air Force)���ݿ��Ѿ��õ����£��������߽����Ȳ������µ�JANAF����(���İ�)[14]��������С���˷���������Mg��Gibbs�ܱ���ʽ����Σ���SGTE ���ݿ���CALPHAD�����õ���Mg�ľ����ȶ�����[15]������0 K��ͬʱ���õ�һԭ������ƽ�沨���Ʒ���[16?22]����hcp-��fcc-��bcc-Mg 0 K�����ܡ���������̬�ܶȵ��������ʣ����CALPHAD��������һԭ������ƽ�沨���Ʒ����͵�һԭ��ͶӰ�Ӳ�����[23?24]�Ľ�����жԱȷ������ҵ�CALPHAD�������һԭ�������IJ��죬̽������Mg�����ȶ��Ե�����ԭ��ͬʱΪMg�Ͻ����ͼ���㼰�ɷ�����ṩ�������ݡ�
1 ԭ���뷽��
1.1 ��С����ԭ��
���ڲ�����Ҫ����Ƶ��������y=f(x)�ϸ��ͨ�����е�ʵ�����ݵ�(xi, yi),��������Ҫ�����ۺ�����xi��ƫ���i=f(xi)?yi���ϸ�ص����㣬���ԣ�Ϊ��ʹ���������ܾ����ط�ӳ�������ݵı仯���ƣ�Ҫ��i����i��2����С��Ϊ�˼��㡢������Ӧ�õķ��㣬һ����ݡ�ƫ��ƽ������С����ԭ��(��Ϊ��С���˷�)��ѡȡ�������,���ɴ���ϵ��ai=(i=0, 1, ???, n)ȷ�������ۺ���Y=f(x, a0, ???, an)ʹ��ƫ��ƽ���ͺ���

����Ȩϵ��wi��ȫȡ1ʱ����Ϊ��Ȩ��С������ϣ���FΪai�ķ����Ժ���ʱ��Ϊ��������С������ϣ���FΪai�����Ժ���ʱ���Ϊ������С�������[25]�����о�����������С������ϣ��Ҽ�Ȩϵ��wiȫȡ1��
�ھ���Ѱ���������y=f(x)����Y=f(x, a0, ???, an)ʱ��һ������ij���������={��0(x), ��1(x), ???, ��n(x)}����x��0��1��???, n�η���������ĺ������ɵĺ����࣬�Թ������ۺ�������

���о�������ȫ��Ԫ��˹��ȥ��ʵ�ֶ�������������[26]��
1.2 Gibbs������ԭ���뷽��
��ȷ������ѧ��������ʽ֮ǰ������JANAF[14]���ݶ�Ԫ��Mg��ת�����ݽ�����������������1���С�
1.2.1 ��ѹ����Cp
��SGTE���������ݿ��У���ѹ���ݣ��ʣ��غ�Gibbs�ܵķֱ�������±�����ʽ[3]��

���о���Ȼ����SGTE���ݿ�ĺ�����ʽ��������С������ϣ���ȷ������Ħ��ͻѹ���ݱ���ʽ(10)�еIJ���c��d������Liquid����ֻ��ȷ������c��
1.2.2 ��H
����SGTE���������ݿ��е���������ʽ����298.15 K��105 Pa�������ȶ�״̬��Ԫ����ֵHSER
Ϊ�ο�̬�ģ��Խ���Mg���ԣ�HSER= ����ˣ�
����ˣ�

������һ��������ȷ��Mg ��hcp��Gibbs�ܱ���ʽ����a������hcp���a��c��dֵ��Liquid���c���Լ���1���۵��¶�ʱ��ת����ֵ��transH������ȷ��Liquid����ֵ����ʽ(11)�еIJ���a��
��1 ����Mg��ת������
Table 1 Transition data of elemental Mg
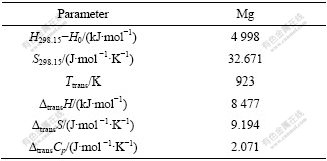
1.2.3 ��S
���Ƶģ����ݱ�1�е�S298.15 K���ɱ���ʽ(12)��

�Ӷ�����ȷ��hcp-Mg��ֵ����ʽ(12)�еIJ���b����������hcp�����b��c��dֵ��Liquid���cֵ���Լ���1���۵��¶�ʱ��ת����ֵ��transS������ȷ��Liquid����ֵ����ʽ(12)�еIJ���b��
1.2.4 ����Ȼ̬������ȷ��
�����Ѿ�ȷ������Ȼ̬hcp-Mg Gibbs�ܱ���ʽ�еIJ���a, b, c��d������SGTE���ݿ��жԹ��ȹ�������T?9��ͶԹ���Һ������T7���CALPHAD�������������ұ��ֶַκ�����ת�����ֵ����ֵ�������ԣ�����ȷ����298.15 K���е�����LiquidҺ��ı���ʽ��Ȼ������Saunders�ľ����ȶ�����[15]���Զ�fcc��bcc������Gibbs�ܱ���ʽ�еIJ�������ȷ����
1.3 ��һԭ������
1.3.1 ��һԭ��
�Բ��Ͻ����������Ļ����Ƕ�����Ѧ���̷���
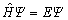 (17)
(17)
ԭ����˵��N �岨�������������п��ܵ���Ϣ��ͨ���ϸ����Ѧ���̷��̣����Եõ���ϵ����������������Ȼԭ���Ͽ���ͨ��������ѧ�Ը���ϵ������⣬�����ڲ�����һ����ϵͳ���ڸ��ӣ������ȡ�����ļͽ��Ʋ�����Ч�ؽ��м��㡣��Ϊ�������ƣ���������3�����轫�����
1) ������۽��ƣ�����������۵�Ѧ���̷��̣�����������۵ĵ����˷��̣�
2) ����?�±���Ĭ���ƣ����ٶ����Ӻͺ˵��˶�����Զ����ģ������Ϳ�����ϵ��Ѧ���̷��̷ֽ�Ϊ�������˶��ķ��������������˶��ķ��̣�
3) �����ӽ��ƣ�������ϵ�е��ӵ��˶�������ÿ��������������ӵ�ƽ���Ƴ��������˶����Ӷ��Ѷ���ӵ�Ѧ���̷��̼�Ϊ��ʽ�ϵĵ����ӷ��̡�
��������3���ٶ�(��Ҫ�Ǻ������ٶ�)��������Ѧ���̷��̵ķ�������Ϊ��ͷ�㷽��(Ab intio)��һ��Ϊ�����ͷ����������������֣�ͨ���ѻ����ܶȷ������۵Ĵ�ͷ���Ϊ��һԭ���������ۻ������ܶȷ�����������ȫ����������ѧ�Ĵ�ͷ�����ۡ�
1.3.2 �ܶȷ�������
HOHENBERG��KOHN[27]��1964��֤������ϵ��̬�ĵ����ܶȷֲ���ȫ������ϵ�����ʣ��Ӷ��춨���ܶȷ�������(Density-functional theory, DFT)�Ļ�����KOHN��SHAM[28]��1965���һ�������Kohn-Sham���̣�

Kohn-Sham���̵Ľ�����־�ŵ�һԭ���ܶȷ������۵���ȫ���������ܶȷ����������������ܶȶ����Dz�������������ϵ��������������ϵ��Ȼ�Ȳ����������ö࣬�ر����ڴ�������ϵʱ��������Եõ�����ļ�
1.3.3 LDA��GGA����
Kohn-Sham����ԭ�����Ǿ�ȷ�ģ�ֻҪ֪����ȷ�������ܶȷ�����ʽ���Ϳ��ɸ÷�������ܶȷֲ������������õ���ϵ�����ʡ�Ȼ������ȫ��ȷ�������ܶȷ�������֪��ֻ��ʹ�ý��Ʒ��������õĽ��������ܶȷ����о����ܶȽ���[29](Local Density Approximation, LDA)�����ݶȽ���[30](Generalized Gradient Approximation, GGA)�����У������ܶȽ���(LDA)�Ļ����뷨�������þ��ȵ��������ܶȺ�����(r)�õ��Ǿ��ȵ������Ľ���-�����������ڸÿ���¿��Ը���EXC[��(r)]�ľ�����ʽ������ͨ�����һ�鵥��������Ч�Ƴ����˶��ķ��̶��õ������ܶȵķֲ��������ڴ˻����ϼ��������й����ԡ��ڹ����ݶȽ���(GGA)�У����������Ʋ���������ܶ��йأ������ܶȺ�����һ�Ͷ�����(�ݶ�)�йأ������ijЩ�����¸����˾����ܶȽ��Ƶļ�������
1.3.4 ƽ�沨���Ʒ���
����ѧ�Ͻ����κ�һ����ѧ�걸����������ΪKohn-Sham���̵ij�ʼ�⣬�����Ҫ��֤���������̲���ɢ����Ѹ����������Ҫ���Ĺ������������ͬ�Ļ������ڽ�Kohn-Sham����ʱ���Ų�ͬ���ŵ��ȱ�㡣�����ܶȷ������۵ij��÷�����ƽ�沨���Ʒ�����������ȫ������Muffintin���[31](Full Potential Linear Muffin-tin Orbital, FPLMTO)��ȫ��������ƽ�沨[32](Full Potential Linear Augmented Plane Wave, FPLAPW)�ȷ�������Щ�����ڼ����м��������κξ���������������ż�ʵ���������ۻ�����ֻҪ�ڼ����������㹻�ߵľ��ȾͿɵõ����ŶȺܸߵļ���� ��[33]��
1.4 ���㷽������Ҫ����
���о�������õ��ǻ����ܶȷ�������(DFT)�ĵ�һ��ԭ������ƽ�沨���Ʒ���, ���еļ��㶼��CASTEP������ɡ������������ܼ����ʹ��GGA ���ƺͳ�������[34]����ÿһԭ������£��Ƚ����еȾ��ȵļ����Ż����ٽ��и߾����Ż��������г��߾����Ż���Ȼ���������ܺ������������ʣ��Ա���PulayӦ��[35]��Ӱ�졣hcp-��fcc-��bcc-Mg k�ռ���������ֱ�Ϊ9��9��6��10��10��10��10��10��10��ƽ�沨���ܽض�����Ϊ380 eV��ͬʱ��Ϊ�˼��ٲ���Ԩ�����ֵ����������������¶�Smearing����[36]�������Ż�ʱ����Gaussian Smearing����[37]��Smearing����Ϊ0.1 eV��
2 ������
2.1 ����ѧ����
2.1.1 Gibbs���������
��������ԭ���ͷ�������SGTE���������ݿ��н���Mg��Gibbs�ܱ���ʽ����������������õ��˱�2�Ľ�����䵥λΪJ/mol��
��2 ��SGTE���������ݿ��н���Mg��Gibbs �ܽ�������������Ľ��
Table 2 Reassessed functions of Gibbs energy of SGTE database of elemental Mg

2.1.2 ��ѹ����Cp�������
��������С���˷�����������JANAF���ݺ�SGTE���������ݿ�õ��ĺ�ѹ����Cp�����˶Աȣ���ͼ1��ʾ��
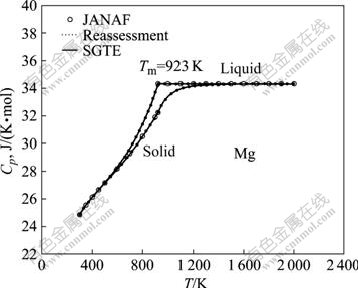
ͼ1 �ɱ���������������SGTE ���ݿ�����Mg �ĺ�ѹ������JANAF���ݵĶԱ�
Fig.1��Comparison of isobaric heat capacity of Mg metals between reassessment calculations in this work, SGTE database and JANAF data
ͼ1�������о���С���˷��������������SGTE���������ݿ�������JANAF����һ�£��ұ��Ľ����SGTE���ݿ����߶��غϣ��������ֺͱȽϣ���ˣ���JANAF����Ϊ�ο�����3��һ�������˶�����������Ķ������
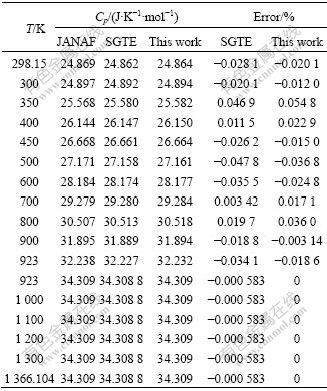
��3 ��������������SGTE ���ݿ�����Mg �ĺ�ѹ������JANAF���ݵĶԱ�
Table 3�� Comparison of isobaric heat capacity of Mg metals between reassessment calculations in this work, SGTE database and JANAF data
2.1.3 �����ȶ������������
���о���SGTE���ݿ���CALPHAD��������SAUNDERS��[15]�ľ����ȶ�����������0 K���������·��������������һԭ�����������жԱȣ�ͼ2��ʾΪ���Ƶľ����ȶ�����ͼ��
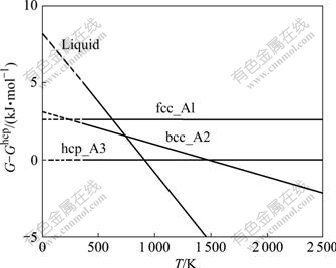
ͼ2 �ɱ�����������0 K�õ��Ľ���Mg �ľ����ȶ�����
Fig.2��Lattice stability parameters extrapolated to 0 K of elemental Mg in this work
2.2 ��һԭ��������
2.2.1 ��������ԭ�����
��4�г����Ż���ľ�������ԭ����������
��4 hcp-��fcc-��bcc-Mg 0 K�����۾�������ԭ�����
Table 4��Theoretical lattice constants of hcp-, fcc- and bcc-Mg at 0 K

2.2.2 ���ܺͽ����
���ܺͽ���ܵļ��������5��ʾ�����н������ָ��0 K��1������ѹ�µĹ����ɻ�̬����ԭ�����������[38]�������ʵĶ��� H=U+PV ֪�����ڹ��������̬��ϵ����������ͬ��ɵ�����dz�С���������Ժ��ԣ������1������ѹ�£�����̬��ϵ�� H �е�PV����Ժ��ԣ���ʱ��H������U������ȣ���ˣ���0 K����ѹ�µĹ����ɻ�̬����ԭ��������������Խ���Ϊ����ܡ�ͬʱ�����Ľ�hcp-Mg�ľ����������㹻Զ(a=1 nm��c=1.633 nm)ʹ�ü����ԭ�Ӽ�������Ϊ�㣬����ʹ�ü���õ�Mgԭ�ӵĹ������ռ����Ϊ1s22s22p6 3s2����ԭ�ӻ�̬�ĵ�����̬��ͬ����ʱ�õ���������Ϊ��̬����ԭ�����ܣ���hcp-��fcc-��bcc-Mg�������������֮��ľ���ֵ��Ϊ����ܡ���5�����˼�������
��5 hcp-��fcc-��bcc-Mg 0K�����ܺͽ����
Table 5 Total energy and cohesive energy of hcp-, fcc- and bcc-Mg at 0 K

2.2.3 ̬�ܶȺ������ռ����
���ӽṹ�IJ����Ǿ����ȶ��Բ���ĸ���ԭ����ˣ����о����㲢�����hcp-��fcc-��bcc-Mg�ļ۲�2p63s2���ӵ���̬�ܶ�(��ͼ3(a))��s̬���ӷ�̬�ܶ�(��ͼ3(b))��p̬���ӷ�̬�ܶ�(��ͼ3(c))��hcp-Mg�ķ�̬�ܶ�(��ͼ3(d))��fcc-Mg�ķ�̬�ܶ�(��ͼ3(e))�� bcc-Mg�ķ�̬�ܶ�(��ͼ3(f))��
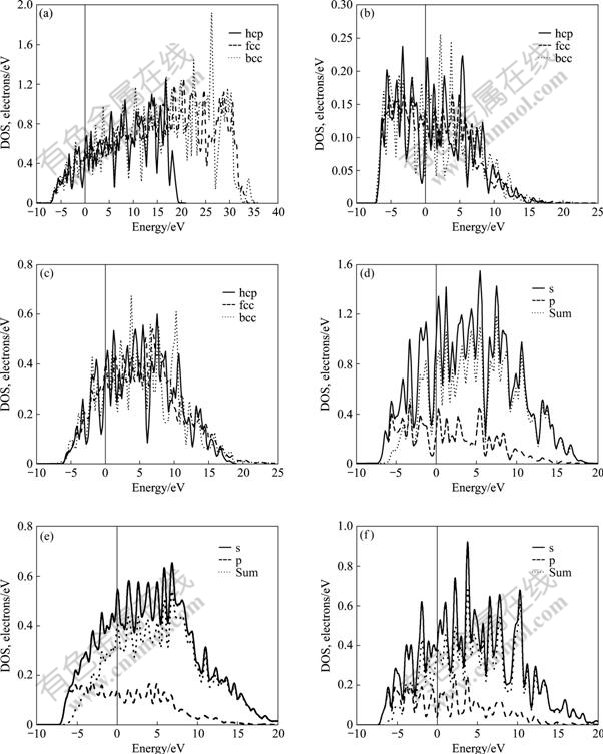
ͼ3 hcp-��fcc-��bcc-Mg��̬�ܶ�
Fig.3��Sum of partial density of state(a), partial density of s state(b), partial density of p state(c) of hcp-, fcc-, bcc-Mg and density of state of hcp-Mg(d), fcc-Mg(e), bcc- Mg(f)
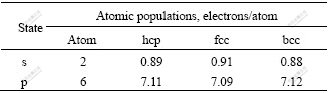
��6 hcp-��fcc-��bcc-Mg �Ĺ������ռ����
Table 6 Atomic populations of hcp-, fcc- and bcc-Mg
3 ����������
3.1 Gibbs���뾧���ȶ�����
��3�����ݶԱȱ��������������Ľ�����۵�923 K��ǰ��11���¶ȵ�����7��������ݱ�SGTE����ȷ���۵��Ժ��6���¶ȵ����SGTE��ȷ��������������ϱ�SGTE������ӽ�JANAF���ݣ��������ȷ��
�����ȶ�����ʵ���������Gibbs�ܣ���������������֮�������ȶ��ԣ��Խ���Mg���ԣ�Ϊfcc-��bcc-Mg���hcp-Mg��Gibbs�ܲ�(G?Ghcp)���¶ȱ仯�Ĺ�ϵ�����ڱ��о�������0 K�ľ����ȶ����⣬��ʱ������Gibbs�ܶ�����
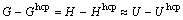 (20)
(20)
��ˣ����Խ�Saunders����CALPHAD����������0K�Ľ�����һԭ������ƽ�沨����(CASTEP)�Լ�ͶӰ�Ӳ�����(VASP)�Ľ�����жԱȣ��������о�����ʵ������������������ڴ����۵ĵ�һԭ������֮���Լ���һԭ��������ͬ����֮��IJ��(G?Ghcp)�Ľ���Ա����7���С�
��7 hcp-��fcc-��bcc-Mg 0K�ľ����ȶ�����
Table7 Lattice stability parameters of hcp-, fcc- and bcc-Mg at 0 K
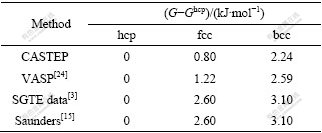
�ɱ�7��֪��1) ���ϵ�һԭ�������õ��ľ����ȶ����������CALPHAD����һ�£���4�ַ�������?Gbcc?hcp��?Gfcc?hcp��0��2) ǰ���ֵ�һԭ�������ľ����ȶ������������CALPHAD����, ��SGTE��Saunders�Ľ��ƫС����bcc���������ƫС��fcc��Ľ������ƫС��3) ��һԭ��CASTEP�����õ���fcc��bcc��ľ����ȶ����������VASP����ƫС��4) ����4�ַ����õ���bcc�����fcc���Gibbs������?Gbcc?fcc����Ϊ1.44��1.37��0.50��0.50 kJ/mol��ǰ���ֵ�һԭ�������ȽϽӽ�������CALPHAD����������ϴ�
3.2 ��������ԭ�����
��8�г��˽���Mg��������ԭ�������һԭ������ƽ�沨���ƺ�ͶӰ�Ӳ������Լ�ʵ��ֵ�ĶԱȽ����
��8 hcp-��fcc-��bcc-Mg 0K�ľ�������ԭ�����
Table 8 Lattice constants and atomic volumes of hcp-, fcc- and bcc-Mg at 0 K

�ɱ�8��֪��1) ����hcp-Mg����һԭ������ƽ�沨���Ʒ���(CASTEP)����ľ�����a��ͶӰ�Ӳ�����(VASP)��ʵ��ֵ��Ҫ�����c/a����ƫС��2)��һԭ������ƽ�沨���Ʒ�������õ���hcp-��fcc-��bcc-Mg��ԭ���������ƫ��ʹhcp-Mg�����c/a��С�������ľ�����a��Ȼ�������������ԭ�������
3.3 ���ܺͽ����
������һ���ܹ㷺�ĸ�������ʵ������������ԭ��Ϊ�ο�̬������̬��ϵ���ܵľ���ֵ��Ϊ�˱��ڱȽ��о�����9������ԭ��Ϊ�ο�̬���Ա����ܺͽ���ܵ����ۼ���ֵ���죺
��9 hcp-��fcc-��bcc-Mg 0 K�����ܺͽ����
Table 9 Total energy and cohesive energy of hcp-, fcc- and bcc-Mg at 0 K
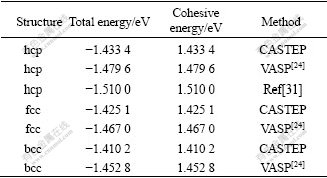
�ɱ�9��֪���ڱ��о����趨�ļ�������£���һԭ������ƽ�沨���Ʒ���(CASTEP)���������(�����)����ֵ��ͶӰ�Ӳ�����(VASP)��ʵ��ֵ��ҪС���뾧������ԭ������Ľ���෴��
3.4 ̬�ܶȺ������ռ����
��ͼ3(a)~(c)����̬�ܶȣ�s��̬�ܶȣ�p��̬�ܶȵĶԱ�֪�����е�s̬��������չ�ֲ���0��?10 eV���䣬��ϱ�6�Ľ��֪������s̬������ת����p̬���ӣ������ͼ3(c)�е�0��?10 eV���������p̬���ӡ�
ͼ3(d)~(f)��һ����0��?10 eV�����̬�ܶȽ����˶Աȡ���ϱ�6�Ľ����֪��1) hcp-Mg(d)s̬���ӵ�̬�ܶ��ڸ�����Ļ������(ͼ�������·����)Ϊ0.89��fcc-Mg(e)��bcc-Mg(f)�ķֱ�Ϊ0.91��0.88������bcc-Mg ÿ��ԭ����1.12��s̬����ת��Ϊp̬���ӣ�ת����Ŀ��࣬hcp-Mg��֮��fcc-Mg���٣�2) ��Fermi������λ�ÿ���hcp-Mg(d)���������ܶȷ�ĵ��У��������Ե�̬�ܶȵ��У�����Ϊ���Ե��ȶ���������fcc-Mg(e)���ڷ�⣬bcc-Mg(f)����һ��һ��������֮�䣬�ṹ���ȶ������ݽ������(�����)�ļ�������bcc��ȶ������������������ٵ�s̬�����йأ�3) �ڽϵ��������䣬̬�ܶ�DOS��˳��ΪDOS(s)��DOS(p)��s̬���ӵ�̬�ܶȴ���̬�ܶȹ�����ϸ������������෴��p̬������������á�
4 ����
1) ���������Ľ�����ӽ����µ�JANAF(���İ�)���ݣ�
2) ��������ƽ�沨�����õ��ľ����ȶ����������CALPHAD�������ƽ���Լ���һԭ��ͶӰ�Ӳ������Ľ����ȫһ�£���?Gbcc?hcp��?Gfcc?hcp��0��
3) ����ƽ�沨���Ʒ�������õ���hcp-��fcc-��bcc-Mg�ľ�������ԭ���������ͶӰ�Ӳ���������������ܾ���ֵ(�����)��ͶӰ�Ӳ�������ƫС��
4) hcp-��fcc-��bcc-Mg�����е�s̬�����ܶ���չ�ֲ���0��?10 eV���䣬���൱һ���ֵ�s̬������ת����p̬���ӣ����У�bcc-Mg��ת����Ŀ��࣬hcp-Mg��֮��fcc-Mg���٣�
5) ̬�ܶȽ������hcpΪ�ȶ��ṹ��fcc��bccΪ���ȶ��ṹ�������ܼ�������һ������fcc�ṹ��bcc���ȶ���̬�ܶ�ͼ�������������Էֱ��ԺͶ������������ȶ��Բ��졣
REFERENCES
[1] KAUFMAN L, BERNSTEIN H. Computer calculation of phase diagram[M]. New York: Academic Press Inc, 1970: 1?58.
[2] SAUNDERS N, MIODOWNIK A P. CALPHAD(Calculation of Phase Diagrams): A comprehensive guide[M]. New York: Pergamon, 1998: 1?66.
[3] DINSDALE A T. SGTE data for pure elements[J]. CALPHAD, 1991, 15(4): 317?425.
[4] SLUITER M H F. Ab initio lattice stabilities of some elemental complex structures[J]. Calphad-Computer Coupling of Phase Diagrams and Thermochemistry, 2006, 30(4): 357?366.
[5] CHRISTENSEN N E, NOVIKOV D L. High-pressure phases of the light alkali metals[J]. Solid State Communications, 2001, 119(8/9): 477?490.
[6] RAHMAN S M M, ALI I, BHUIYAN G M. Phase stability of alkali metals under pressure: Perturbative and non-perturbative treatments[J]. International Journal of Modern Physics B, 2002, 16(32): 4847?4864.
[7] GARCES J E, GRAD G B, GUILLERMET A F. Theoretical study of the structural properties and thermodynamic stability of the omega phase in the 4d-transition series[J]. Journal of Alloys and Compounds, 1999, 289(1/2): 1?10.
[8] GRAD G B, BLAHA P, LUITZ J. Electronic structure and chemical bonding effects upon the bcc to Omega phase transition: Ab initio study of Y, Zr, Nb, and Mo[J]. Physical Review B, 2000, 62(19): 12743?12753.
[9] GUO G Y, WANG H H. Calculated elastic constants and electronic and magnetic properties of bcc, fcc, and hcp Cr crystals and thin films[J]. Physical Review B, 2000, 62(8): 5136?5143.
[10] SIN'KO G V, SMIRNOV N A. Ab initio calculations of elastic constants and thermodynamic properties of bcc, fcc, and hcp Al crystals under pressure[J]. Journal of Physics-Condensed Matter, 2002, 14(29): 6989?7005.
[11] SKRIVER H L. Crystal structure in one-electron theory[J]. Physical Review B, 1985, 31(4): 1909?1923.
[12] ������, ���, ��ѧ��. ����-Mn�ĵ��ӡ���̬���Ժʹ�����ṹ[J]. ����ѧ��, 2002, 38(12): 1251?1256
CHEN X Q, LI H L, DING X Y. Electronic structure, ground properties and magnetic ordering for pure ��-Mn[J]. Acta Metallurgica Sinica, 2002, 38(12): 1251?1256.
[13] MISHIN Y, MEHL M J, PAPACONSTANTOPOULOS D A, VOTER A F, KRESS J D. Structural stability and lattice defects in copper: Ab initio, tight-binding and embedded-atom calculations[J]. Physical Review B, 2001, 63(22): 224106?224121.
[14] CHASE M W. NIST-JANAF Thermochemical Tables (Fourth Edition Part I) [M]. Gaithersburg: National Institute of Standards and Technology, 1998: 1005?1009.
[15] SAUNDERS N, MIODOWIK A P, DINSDALE A T. Metastable lattice stabilities for the elements[J]. Calphad-Computer Coupling of Phase Diagrams and Thermochemistry, 1988, 12(4): 351?374.
[16] SRIVASTAVA G P, WEAIRE D. The theory of the cohesive energies of solids[J]. Advances in Physics, 1987, 36(4): 463?517.
[17] BACHELET G B, HAMANN D R, SCHLUTER M. Pseudopotentials that work: From H to Pu[J]. Physical Review B, 1982. 26(8): 4199?4228.
[18] LIN J S, QTEISH A, PAYNE M C, HEINE V. Optimized and transferable nonlocal separable ab initio pseudopotentials[J]. Physical Review B, 1993, 47(8): 4174?4180.
[19] VANDERBILT D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 1990, 41(11): 7892?7895.
[20] MILMAN V, WINKLER B, WHITE J A, PICKARD C J, PAYNE M C, AKHMATSKAYA E V, NOBES R H. Electronic structure, properties and phase stability of inorganic crystals: A pseudopotential plane-wave study[J]. International Journal of Quantum Chemistry, 2000, 77(5): 895?910.
[21] FRANCIS G P, PAYNE M C. Finite basis set corrections to total energy pseudopotential calculations[J]. Journal of Physics: Condensed Matter, 1990, 29(19): 4395?4404.
[22] MILMAN V, LEE M H, PAYNE M C. Ground-state properties of CoSi2 determined by a total-energy pseudopotential method[J]. Physical Review B, 1994, 49(23): 16300?16308.
[23] BL?CHL P E, JEPSEN O, ANDERSEN O K. Improved tetrahedron method for Brillouin-zone integrations[J]. Physical Review B, 1994, 49(23): 16223?16233.
[24] WANG Y, CURTAROLO S, JIANG C, ARROYAVE R, WANG T, CEDER G, CHEN L Q, LIU Z K. Ab initio lattice stability in comparison with CALPHAD lattice stability[J]. CALPHAD, 2004, 28(1): 79?90.
[25] ����. �ִ�Ӧ����ѧ�ֲ�?��������ֵ������[M]. ����: �廪��ѧ������, 2005: 15?52.
MA Zhen-hua. Handbook of modern applied mathematics: Calculations and numerical analysis[M]. Beijing: Tsinghua University Press, 2005: 15?52.
[26] �״���, ���Ʊ�, ���з�. ���㷽��[M]. ����: �㽭��ѧ������, 1996: 77?86.
YI D Y, SHEN Y B, LI Y F. Calculation methods[M]. Hangzhou: Zhejiang University Press, 1996: 77?86.
[27] HOHENBERG P, KOHN W. Inhomogeneous electron gas[J]. Physical Review B, 1964, 36(3): 864?871.
[28] KOHN W, SHAM L J. Self-consistent equations including exchange and correlation effects[J]. Physical Review A, 1965, 140 (4): 1133?1138.
[29] PERDEW J P, WANG Y. Accurate and simple analytic representation of the electron gas correlation energy[J]. Physical Review B, 1992, 45(23): 13244?13249.
[30] PERDEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865?3868.
[31] ANDERSEN O K. Linear methods in band theory[J]. Physical Review B, 1975, 12(8): 3060?3083.
[32] BLAHA P, SCHWARZ K, SORANTIN P, TRICKEY S B. Full-potential, linearized augmented plane wave programs for crystalline systems[J]. Computer Physics Communications, 1990, 59(2): 399?415.
[33] DEVANATHAN R, CORRALES L R, WEBER W J, CHARTIER A, MEIS C. Molecular dynamics simulation of defect production in collision cascades in zircon[J]. Nuclear Instruments and Methods in Physics Research, Section B: Beam Interactions with Materials and Atoms, 2005, 228(1/4): 299?303.
[34] JOHN P P, WANG Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B, 1992, 45(23): 13244?13249.
[35] PULAY P. Convergence acceleration of iterative sequences. the case of scf iteration[J]. Chemical Physical Letters, 1980, 73(2): 393?398.
[36] MERMIN N D. Thermal properties of the inhomogeneous electron gas[J]. Physical Review A, 1965, 137: 1441?1443.
[37] METHFESSEL M, PAXTON A T. High-precision sampling for Brillouin-zone integration in metals[J]. Physical Review B, 1989, 40(6): 3616?3621.
[38] KITTEL C. Solid state physics[M]. New York: John Wiley and Sons Inc, 1976, 55.
[39] WINTER M. Webelements[OL]. http://www.webelements.com/. The University of Sheffield and Webelements Ltd, 2006.
������Ŀ����������ʿ���½�ʦ����(20070533118), ������Ȼ��ѧ����(50471058, 50271085), ���ϴ�ѧ��ʿ���������
�ո����ڣ�2008-03-06�������ڣ�2008-06-24
ͨѶ���ߣ��ջԽ�����ʿ���绰��0731-8830163��Tax��0731-8876692��E-mail: thj@mail.csu.edu.cn
(�༭ ��ѧ��)

