����-̼�Ȼ�ԭ���Ϸ��Ʊ����ʯ�������
�� ��, ���º�, ���Ȩ, ��־��, ������
(���ϴ�ѧ ұ���ѧ�빤��ѧԺ, ��ɳ 410083)
ժ Ҫ: ��FeSO4��7H2O, NH4H2PO4��H2O2Ϊ��ʼԭ��, ͨ��Һ������Ƶ�ǰ����FePO4, Ȼ��ͨ��̼�Ȼ�ԭ�õ�LiFePO4/C�� X���������ɨ��羵��������: 560, 600, 700��800��ϳɵ���Ʒ��ΪLiFePO4/C, LiFePO4����������ϳ��¶ȵ����߶�������, 560��ϳɲ��ϵĿ��������ֲ���0.3~0.4��m֮��; ��800��ϳɲ��ϵĿ���������ﵽ0.6~0.7��m, ��Ӧʣ���̼��ֱ�ӷֲ���LiFePO4����֮��, �������������ӵ����ʡ� 560����Ʒ�ڷŵ籶��Ϊ0.1Cʱ���״ηŵ������Ϊ151mA��h/g(0.1C), �����ŵ籶�ʴﵽ1Cʱ, �ŵ������Ϊ129mA��h/g, �Ҿ������õ�ѭ�����ܡ�
�ؼ���: LiFePO4; Һ�����; ̼�Ȼ�ԭ; ǰ���� ��ͼ�����: TM912.9
���ױ�ʶ��: A
Synthesis of lithium iron phospho-olivines by aqueous precipitation and carbothermal reduction
ZHANG Bao, LI Xin-hai, ZHU Bing-quan,WANG Zhi-xing, GUO Hua-jun
(School of Metallurgical Science and Engineering,Central South University, Changsha 410083, China)
Abstract: LiFePO4 was prepared by carbothermal reduction of FePO4 which was synthesized by aqueous precipitation from FeSO4��7H2O and NH4H2PO4 and hydrogen peroxide as the oxidizing agents. Samples were characterized by XRD and SEM. It is shown that the pure and homogenous LiFePO4 was successfully synthesized at 560, 600, 700 and 800��, with average particle sizes about 0.3-0.4��m for the material synthesized at 560�� and 0.6-0.7��m at 800��. The residual carbon during processing was coated on LiFePO4, resulting in the enhancement of the material��s electronic properties. The discharge capacity of the material synthesized at 560�� is 151mA��h/g at 0.1C rate and 129mA��h/g at 1C rate.
Key words: LiFePO4; aqueous precipitation; carbothermal reduction; precursor
1997��Padhi��[1]������LiFePO4���г�ŵ�����, �����۱�����Ϊ170mA��h/g, �ŵ�ƽ̨Ϊ3.4V, �������õ�ѭ������[2]�����ȶ�����[3, 4], �Ҿ������� �۸���˵��ص�, ʹ��������Ϊ��һ�����������ϡ�
�����ֲ��ϵ�ijЩȱ���谭������ʵ��Ӧ��[5-10]: һ�Ǻϳ���Fe2+��������Fe3+, ���õ������LiFePO4; ����LiFePO4�����Բ�, ���������ŵ����ܲ ���е��о�ͨ�����¼����������LiFePO4������[11-17]: ���ö�������������Fe2+; �ϳ�С������LiFePO4�������ӵ���ɢ����; �ӵ絼������ߵ絼�ʡ� ����ϳɷ����Բ��ϵ����ܼ�ʵ��Ӧ���кܴ��Ӱ��, ���еķ�����Ҫ�й��෨[18]�� ˮ�ȷ�[19]�� �ܽ�-������[20]��̼�Ȼ�ԭ��[21]�� ̼�Ȼ�ԭ������Ϊ�DZȽ���ǰ;��һ�ַ���, ��Ϊ�ϳɵIJ��ϵ绯ѧ���ܺ���ʵ����������[22]�� Mich��[21]�Է�������FePO4��LiOHΪԭ��, �۱�ϩΪ��ԭ��, �ϳɵIJ�����0.1C��0.5C�������״ηŵ�������ֱ�Ϊ160mA��h/g��146.5mA��h/g�� �������߲��øĽ���̼�Ȼ�ԭ��, ����FeSO4��7H2O��NH4H2PO4Ϊԭ��, ����Һ��������Ʊ�FePO4ǰ����, Ȼ��ǰ���塢 Li2CO3������̼�ڻ�Ͼ���, �ڸ����»�ԭ�ϳ�LiFePO4��
1 ʵ��
1.1 LiFePO4/C���Ʊ�
��ȡ��Ħ����FeSO4��7H2O��NH4H2PO4(��ѧ��)��2����ƿʢ��, ����ȥ����ˮ�����Һ�� ��2����Һ��ϲ��ڷ�Ӧ���в�ͣ���跴Ӧһ��ʱ��, Ȼ�����ŨH2O2(Ũ��>30%), ��Ӧ�������д���FePO4��ɫ���������� ��Ӧ������������ķ�����ϴ��, ��ϴ�Ӻ���˱���ɡ� ����ɵ��������� ̼��﮼�����̼�ڰ���ѧ�����Ȼ�Ͼ���(̼����10%), ��Ar���ı����·ֱ���500, 560, 600, 700��800��������12h, �õ�LiFePO4/C, �ֱ��ΪS1, S2, S3, S4��S5, ����̼������������̼�����ֱ�Ϊ10.21%, 10.10%, 10.15%, 9.91%��9.94%��
1.2 ����
���� CS800����̼������(Eltar��˾, �¹�)�Ժϳɲ�����̼�ĺ������з����� TG-DTA�Dz�������������SDTQ600, �¶ȴ�20������760��, �����ٶ�Ϊ10��/min, �Ԧ�-Al2O3��Ϊ�α�� ��X����������(Rint-2000, Rigaku)��ɨ��羵(JEOL, JSM-5600LV)�о���Ʒ�ľ���ṹ�ͱ�����ò��
1.3 �����װ�Ͳ���
ʵ��������Ƭ��m(LiFePO4/C)��m(̼��)��m((PVDF)(��ƫ����ϩ))=8��1��1�ı�����Ͼ��Ⱥ�ͿƬ�Ƴ�, �缫Ƭ��ֱ��Ϊ14mm, ���а���LiFePO4��������ԼΪ5mg���ҡ� ���Ʊ��õ�����Ƭ��120����ո���10h�� �����Ƭ��Ϊ����, 1mol/L LiFP6/EC+DMC (�����1��1)Ϊ���Һ, ������������װ��CR2025�Ϳ�ʽ��ء� �������������Dz��Ե������, ��ŵ��ѹ������4.1~2.3V��
2 ���������
ͼ1��ʾΪǰ������������TG-DTA���ߡ� ��DTA���߿��Կ�����, ��150�����ҳ������Ե����ȷ�, ͬʱ��������������ʧ, �˷�ӦΪǰ����ʧȥ�ᾧˮ�Ĺ���; ����580���710�����ҳ���2�����ȷ����û����������ʧ, ����ֱܷ��Ӧ��������������ת��Ϊ������, ���Ӧ�����ת��Ϊ�¾��͵�2������[5]�� ��TG���߿��Կ���, ��20~500��֮��, ǰ����������ʧΪ19%, �����ظ���صζ�ǰ�������ĺ���, ����京��Ϊ29.88%, ��Щ����������[23]����������������2���ᾧˮ���Ǻϡ� ͼ2��ʾΪǰ����FePO4��SEM�� ���Կ���, �ϳɵ�ǰ�������������״, �ֲ��dz�����, ����С(Լ0.1��m), �ȱ������
ͼ3��ʾΪFePO4��2H2O, Li2CO3��C����ϵ�TG-DTA����(Ħ����Ϊ2��1��3.92)�� ��DTA
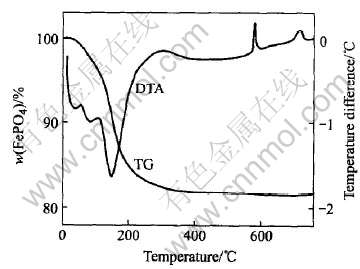
ͼ1 FePO4��TG-DTA����
Fig.1 TG-DTA curves for precipitated FePO4

ͼ2 FePO4��ɨ��羵��
Fig.2 SEM image of FePO4

ͼ3 FePO4��2H2O, Li2CO3��C����ϵ�TG-DTA����
Fig.3 TG-DTA curves for mixture of precursor, Li2CO3 and C
���߿�֪, ��150��������һ����Ӧ���շ�, �˷�ӦΪFePO4��2H2Oʧȥ�ᾧˮ�� ��545���660����2�����ȷ�, ��Ӧ��TG�߸պ�Ҳ��2��������ʧ��, ��Ӧ�ķ�ӦΪ

������Ϊ̼������Ӧ��2�����, ��:

�������J��˹������ͼ[24]���Կ���, ���¶ȵ���650��ʱ, ��Ӧ(3)�ļ���˹�����ܱȷ�Ӧ(4)�ļ���˹������С, �����¶ȸ���650��ʱ��Ӧ(4)�ļ���˹��������Խ�С, Ҳ����˵�������¶ȵ���650��ʱ����CO2, �����¶ȸ���650��������CO��

ͼ4 ��ͬ�¶��ºϳ�LiFePO4��X����������
Fig.4 XRD patterns of LiFePO4/C prepared at different temperatures
ͼ4��ʾΪ��ͬ�¶��ºϳɵ�LiFePO4/C��X���������ס� ���Կ���, ��ƷS2~S5û���κ����ʷ�, Ϊ��LiFePO4, ���źϳ��¶ȵ�����, ��������б���� 500��ϳɵ���Ʒ�к����Ե����ʷ�, ������ΪLi3Fe2(PO4)3�� ͼ5��ʾΪ��ͬ�¶��ºϳ�LiFePO4/C��SEM�� ���Կ���, �д�ϸ�������, ���н�ϸ��Ϊ̼��, �ϴֵ�ΪLiFePO4, ��LiFePO4�Ŀ������źϳ��¶ȵ��������� 560���ºϳɵ�������﮿����ֲ���0.3~0.4��m֮��, ���¶����ߵ�800��, ��������0.6~0.7��m�� ����Prosini��[25]���о�, ��̼��Ϊ10%ʱ, LiFePO4/C���ϵ��ӵ�����Ϊ1.7��10-3 S/cm, �ȴ���LiFePO4�ĵ��ӵ�����(10-10 S/cm)����˽�7���������� Ҳ����˵��̼�����������LiFePO4�ĵ��ӵ�����, ����SEM����Կ���, LiFePO4����Χ�ֲ��ŷ�Ӧʣ���̼��, �������������߲��ϵĵ��ӵ����ʡ�
ͼ6��ʾΪ��ͬ�¶��ºϳɵ�LiFePO4/C��0.1C�������״γ�ŵ����ߡ� ��ŵ�Ľ�ֹ��ѹΪ4.1~2.3V�� ��ͼ�п��Կ���, ���źϳ��¶ȵĽ���, �ŵ�������������, ��t=560��ʱ, �ŵ�������ﵽ151mA��h/g, ��t=800��ʱ, �ŵ������ֻ��100mA��h/g(t=500��ʱ������С, ����û���г�)�� �����������źϳ��¶ȵ�����, ��Ʒ�������, ����Andersson��[18]������, ���Ƕ��/�ѳ�������, ������Խ��Ŀ���, �������ɢ·�̾�Խ��, �ڿ������ĸ����Ļ�������Ҳ��Խ�����á�
ͼ7��ʾΪ560��ϳɵ�LiFePO4/C�ڲ�ͬ�����µij�ŵ�����(����0.2C���ʳ��)�� ���Կ���, ���ŷŵ����������, ��صķŵ��������С, ��0.2C���ʷŵ�ʱ, ����Ϊ148mA��h/g, ����1C���ʷŵ�ʱ, ��������129mA��h/g�� ͬʱҲ���Կ������ų�ŵ����������, �伫��Ҳ��Ӧ���� ͼ8��ʾΪ560���ºϳɵ�LiFePO4��0.5C��1C�ŵ籶���µ�ѭ���������ߡ� ���Կ���, �ڸ������ºϳɵIJ����ڴ�����ŵ��¾������õ�ѭ�����ܡ�

ͼ5 ��ͬ�¶��ºϳ�LiFePO4��SEM��
Fig.5 SEM images of LiFePO4/C prepared at different temperatures
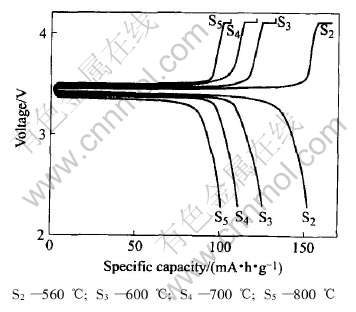
ͼ6 ��ͬ�¶��ºϳ�LiFePO4/C��0.1C�����µij�ŵ�����
Fig.6 Voltage profiles for LiFePO4/C prepared at different temperatures recorded at 0.1C
���ô˷����ϳɵIJ���, С�����ŵ�������Ե�������[21]�������ķŵ������(Ϊ�������,����[21]���ϳɵIJ��ϼ�ΪR����), ���������и��õ�ѭ�����ܡ� R������0.3C���ʷŵ�ʱ, ����25��ѭ����, �ŵ������û��˥��, ����0.5C�����¾���25��ѭ����, R���ϵķŵ��������146.5mA��h/g˥����142.5mA��h/g, ˥����Ϊ2.73%; ���������ϳɲ�����0.5C�����¾���30��ѭ����, ������ȴ��137mA��h/g������139 mA��h/g�� ����[21]û�����������ŵ�����(1C), �������ϳɵIJ�����1C�����¾���30��ѭ����, �ŵ������˥���ʽ�Ϊ1.55%(��129mA��h/g˥����127mA��h/g)�� ͬʱ, ����[21]�����õ�ԭ�������
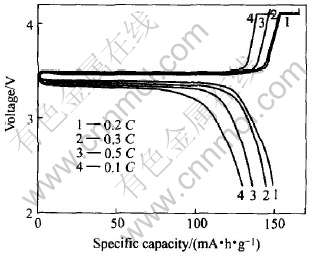
ͼ7 560�������ºϳɲ��ϵı�������
Fig.7 Voltage profiles for LiFePO4 prepared at 560�� recorded at different discharge rates(charge rate 0.2C)

ͼ8 560�������ºϳɲ��ϵ�ѭ������
Fig.8 Electrochemical cycling performance for LiFePO4/C prepared at 560��
REFERENCES
[1]Panhi A K, Nanjundaswamy K S, Goodenpugh J B. Phospho-olivines as positive-electrode materials for rechargeable lithium batteries[J]. J Electrochem Soc, 1997, 144: 1188-1194.
[2]Anna S A, Beata K, Lennart H, et al. Lithium extraction/insertion in LiFePO4: an X-ray diffraction and Mossbauer spectroscopy study[J]. Solid State Ionics, 2000, 133: 41-52.
[3]Macneil D D, Lu Z, Chen Z, et al. A comparison of electrode/electrolyte reaction at elevated temperature for various Li-ion battery cathodes[J]. J Power Sour, 2002, 108: 8-14.
[4]Takahashi M, Tobishima S, Takei K, et al. Reaction behavior of LiFePO4 as a cathode material for rechargeable lithium batteries[J]. Solid State Ionics, 2002, 148: 283-289.
[5]Prosini P P, List M, Scaccia S, et al. Synthesis and characterization of amorphous hydrated FePO4 and its electrode performance in lithium batteries[J]. J Electrochem Soc, 2002, 149: A297-A301.
[6]Prosini P P, List M, Zane D. Determination of the chemical diffusion coefficient of lithium in LiFePO4[J]. Solid State Ionics, 2002, 148: 45-51.
[7]Rissouli K, Benkhouja K, Ramos-Barrado J R, et al. Electrical conductivity in lithium orthophosphates[J]. Mater Sci Eng B, 2003, B98: 185-189.
[8]Zhou F, Kang K, Maxisch T, et al. The electronic structure and band gap of LiFePO4 and LiMnPO4[J]. Solid State Communications, 2004, 132: 181-186.
[9]Yamada A, Chung S C, Hinokuma K. Optimized LiFePO4 for lithium battery cathodes[J]. J Electrochem Soc, 2001, 148: A224-A229.
[10]Zane D, Carewska M, Scacia S, et al. Factor affecting rate performance of undoped LiFePO4[J]. Electrochimica Acta, 2004, 49: 4259-4271.
[11]Bauer M E, Bellitto C, Pasoualli M, et al. Versatile synthesis of carbon-rich LiFePO4 enhancing its electrochemical properties[J]. Electrochem and Solid State Lett, 2004, 7: A85-A87.
[12]Cho T, Chung H. Synthesis of olivine-type LiFePO4 by emulsion-drying method[J]. J Power Sour, 2004, 133: 272-276.
[13]Park K S, Son J T, Chuang H T, et al. Surface modification by silver coating for improving electrochemical properties of LiFePO4[J]. Solid State Communications, 2004, 129: 311-314.
[14]Prosini P P, Carewska M, Scaccia S, et al. Long-term cyclability of nanostructured LiFePO4[J]. Electrochimica Acta, 2003, 48: 4205-4211.
[15]Chuan L, James A. Carbonization and activation of sol-gel derived carbon xerogels[J]. Carbon, 2000, 38: 849-861.
[16]Doeff M M, Hu Y, Mclaron F, et al. Effect of surface carbon structure on the lectrochemical performance of LiFePO4[J]. Electrochem and Solid State Lett, 2003, 6: A207-A209.
[17]Herstedt M, Stjerndahl M, Nyten A, et al. Surface chemistry of carbon-treated LiFePO4 particles for Li-ion battery cathodes studied by PES[J]. Electrochem and Solid state Lett, 2003, 6: A202-A206.
[18]Andersson A S, Thomas J O. The source of first-cycle capacity loss in LiFePO4[J]. J Power Sources, 2001, 97/98: 498-502.
[19]Yang S, Zavalij P Y, Whittngham M S. Hydrothermal synthesis of lithium iron phosphate cathodes[J]. Electrochemistry Communications, 2001, 3: 505-508.
[20]Hu Q, Doeff M M, Kostecki R, et al. Electrochemical performance of sol-gel synthesized LiFePO4 in lithium batteries[J]. J Electrochem Soc, 2004, 151: A1279-A1285.
[21]Mich H, Cao G S, Zhao X B. Low-cost, one-step process for synthesis of carbon-coated LiFePO4 cathode[J]. Materials Letters, 2005, 59: 127-130.
[22]Barker J, Saidi M W, Swoyer J L. Lithium Iron(��) phospho-olivines prepared by a novel carbothermal reduction method[J]. Electrochem and Solid state Lett, 2003, 6(3): A53-A55.
[23]Scaccia S, Carewska M, Wisniewski P, et al. Morphological investigation of sub-micron FePO4 and LiFePO4 particles for rechargeable lithium batteries[J]. Materials Reserch Bulletin, 2003, 38: 1155-1163.
[24]����˵. ��ɫұ��ԭ��[M]. ����: ұ��ҵ������, 1992. 38.
FU Chong-yue. The principle of non-ferrous metallurgy[M]. Beijing: Metallurgical Industry Press, 1992. 38.
[25]Prosini P P, Zane D, Pasquali M. Improved electrochemical performance of a LiFePO4-based composite cathode[J]. Electrochimica Acta, 2001, 46: 3517-3523.
(�༭�°���)
������Ŀ: ������Ȼ��ѧ����������Ŀ(50302016)
�ո�����: 2005-09-28; ������: 2006-02-21
ͨѶ����: �� ��, ��ʿ; �绰: 0731-8836357; E-mail: zhangb@qianlong.com

