����ʯ�Ͱ���ʯ�����µĻ��������������������ѧģ��ͱ������
��Դ�ڿ����й���ɫ����ѧ��(Ӣ�İ�)2018���7��
�������ߣ�Ehsan BIDARI Valeh AGHAZADEH
����ҳ�룺1433 - 1443
�ؼ��ʣ����������̼������ʯ�������ͽ��
Key words��pyrite; alkaline leaching; carbonate gangue; Carlin gold deposits
ժ Ҫ��ͨ����Ъ�������飬��ϻ�ѧģ�⡢��Һ������������������о�����ʯ�Ͱ���ʯ�Ի�����������Ӱ�졣��80 ��C��p(O2)=1.013��105 Pa��pH>13�������½��н������顣����������ڷ���ʯ�Ͱ���ʯ���������£��������ڼ��е��������½������з���ʯ��Ӱ�����FE-SEM/EDS������������������溬�и�(����ʯ����ʱ)��þ(����ʯ����ʱ)�����ǻ�����������½���ԭ���ݻ�ѧģ��������һ�������γ�������̼���ο���IJ����ܽ⣬��Ҫ�ܽ���ҺpHֵ���¶ȵ�Ӱ�졣XRD��FTIR���������������Ϊ�ƻ�þ����������о������֣��ڷ���ʯ���ڵ�����£�����㱡�������������ģ����ڰ���ʯ���������£������Ⱥ��ֲ�������
Abstract: The effect of calcite and dolomite on the oxidation of pyrite was studied using batch-leaching experiments complemented with chemical modeling, solution analysis and solid characterization techniques. Leaching tests conducted at 80 ��C and p(O2)=1.013��105 Pa and pH>13 showed that pyrite alkaline oxidation rate decreased in the presence of both calcite and dolomite, while more detrimental effect was observed in the case of calcite. FE-SEM/EDS analysis exhibited a surface layer containing Ca (in the case of calcite) and Mg (in the case of dolomite) on the pyrite surface, which justified the slowdown in pyrite leaching rate. This surface layer was formed due to partial dissolution of carbonate minerals, which was affected dominantly by the pH and temperature of the leaching solution based on the chemical modeling data. The surface layer was characterized as Ca or Mg hydroxide using XRD and FTIR analysis. It was also found that this layer was thin and continuous in the case of calcite while it was thick and discontinuous in the presence of dolomite.
Trans. Nonferrous Met. Soc. China 28(2018) 1433-1443
Ehsan BIDARI, Valeh AGHAZADEH
Department of Mineral Processing, Faculty of Mining Engineering, Sahand University of Technology, Tabriz, Iran
Received 2 June 2017; accepted 4 October 2017
Abstract: The effect of calcite and dolomite on the oxidation of pyrite was studied using batch-leaching experiments complemented with chemical modeling, solution analysis and solid characterization techniques. Leaching tests conducted at 80 ��C and p(O2)=1.013��105 Pa and pH>13 showed that pyrite alkaline oxidation rate decreased in the presence of both calcite and dolomite, while more detrimental effect was observed in the case of calcite. FE-SEM/EDS analysis exhibited a surface layer containing Ca (in the case of calcite) and Mg (in the case of dolomite) on the pyrite surface, which justified the slowdown in pyrite leaching rate. This surface layer was formed due to partial dissolution of carbonate minerals, which was affected dominantly by the pH and temperature of the leaching solution based on the chemical modeling data. The surface layer was characterized as Ca or Mg hydroxide using XRD and FTIR analysis. It was also found that this layer was thin and continuous in the case of calcite while it was thick and discontinuous in the presence of dolomite.
Key words: pyrite; alkaline leaching; carbonate gangue; Carlin gold deposits
1 Introduction
Pyrite (FeS2) is the most common sulfide in the Earth��s surface region. Not only is the mineral ubiquitous, but the reactivity of pyrite is of central importance in a devastating environmental issue known as acid mine drainage (AMD) [1]. Aside from environmental concerns, undesirable association of pyrite with minerals of economic value such as sphalerite, chalcopyrite and galena, and precious metals such as gold necessitates costly separation processes such as leaching and flotation [2,3]. Specifically, auriferous pyrite with gold finely disseminated in its matrix is rather common. However, pyrite is relatively stable in cyanide, so that the encapsulated gold particles cannot be recovered during cyanidation after normal grounding processes. This kind of gold ores need to be submitted to some forms of oxidation treatment before cyanidation process [4-6]. Considering two literature review articles on pyrite oxidation behavior [1,2], it can be seen that most of researches to date have focused on acidic oxidation of pyrite. Nonetheless, alkaline oxidation can be a possible alternative to acidic oxidation specifically in the case of high carbonate refractory gold ores to avoid high acid consumption and also to suppress elemental sulfur formation (which is responsible for agglomeration and gold losses). Additionally, oxidizing the gold encapsulating pyrite in the alkaline solution improves the oxidative power of oxygen in comparison to acidic solution by reducing CO2 production. Moreover, there is a new trend in gold hydrometallurgy aiming to develop a new method in order to reach simultaneous dissolution of gold with in situ formed thiosulfate during pyrite oxidation [7-9] which needs to be conducted under alkaline condition. Pyrite associated with carbonate minerals is rather common specifically in gold deposits [10,11]. For example, in Carlin type gold deposits, calcite and dolomite are the principal minerals of the host rocks and the ore consisted mainly of pyrite [12-14]. While alkaline leaching was proposed for auriferous pyrite oxidation in such deposits [11,15-18], there is no available study concerning the effect of these gangue minerals on the alkaline oxidation of pyrite. The aim of the present investigation is 1) to carry out a brief thermodynamic and kinetic investigation on the dissolution behavior of pyrite, calcite and dolomite in the alkaline solution, 2) to determine the effect of the presence of calcite and dolomite on pyrite alkaline oxidation experimentally and 3) to apply solution analysis and solid characterization in order to suggest the possible role of these gangue minerals on pyrite oxidation.
2 Thermodynamic and kinetic investigation
It was found that the pyrite leaching was mainly controlled by chemical reaction [19]. It was also reported that pyrite is stable in a relatively broad range from approximately pH 1 to pH 13. The potential range for pyrite stability is from -0.6 to 0.3 V. It is interesting to note that above pH 13, pyrite is no longer thermodynamically stable at any potential. The rest potential of pyrite is about 0.3 V at pH 9.2, 0.2 V at pH 10 and -0.07 V at pH 11. Thus, normal oxidants such as oxygen can oxidize pyrite in alkaline media, yielding iron hydroxides and sulfate ions [20]. Accordingly, the leaching tests of this study were conducted at pH above 13 and using oxygen as oxidation agent. At high pH values, the iron exists in the form of hydrated iron oxide species. The iron hydroxide may form goethite (��-FeOOH), and then hematite (��-Fe2O3), as the water of hydration is removed at higher temperatures. In hydroxide medium, hematite is the main product of iron oxidation, while in contrast the main constituent is ferrihydrite in carbonate medium[17]. Subsequently, based on these fundamental studies the overall stoichiometry for aqueous oxidation of pyrite by molecular oxygen in alkaline solutions may be represented as
4FeS2+15O2+16OH- 4Fe(OH)3+
4Fe(OH)3+ +2H2O (1)
+2H2O (1)
This overall reaction is made up of the two half reactions:
FeS2+11H2O��Fe(OH)3+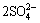 +19H++15e (2)
+19H++15e (2)
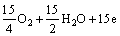 ��15OH- (3)
��15OH- (3)
The presence of other species such as 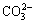 or Ca2+, Mg2+ may affect the reaction mechanism of pyrite oxidation [21]. This may happen due to calcite or dolomite dissolution. The solubility products of sparingly soluble calcium and magnesium compounds at 25 ��C follow the order of CaMg(CO3)2(s)(10-17.2) << Mg(OH)2(s) (10-11.3) << CaCO3(s) (10-8.1) < Ca(OH)2(s) (10-5.4) < MgCO3(s) (10-4.7) < CaSO4(s) (10-4.3) [22-24]. It is clear that the variation in solution parameters such as temperature and pH can affect the precipitation behavior of these species. Calcite solubility as a function of pH and temperature was studied by several researchers [25-29]. Accordingly, Ca(OH)2(s), Ca(OH+)(aq) and (aq) were reported as the favorable calcite dissolution species under the experimental conditions prevailing in the current study (pH>13, T=80 ��C) (Eq. (4)).
or Ca2+, Mg2+ may affect the reaction mechanism of pyrite oxidation [21]. This may happen due to calcite or dolomite dissolution. The solubility products of sparingly soluble calcium and magnesium compounds at 25 ��C follow the order of CaMg(CO3)2(s)(10-17.2) << Mg(OH)2(s) (10-11.3) << CaCO3(s) (10-8.1) < Ca(OH)2(s) (10-5.4) < MgCO3(s) (10-4.7) < CaSO4(s) (10-4.3) [22-24]. It is clear that the variation in solution parameters such as temperature and pH can affect the precipitation behavior of these species. Calcite solubility as a function of pH and temperature was studied by several researchers [25-29]. Accordingly, Ca(OH)2(s), Ca(OH+)(aq) and (aq) were reported as the favorable calcite dissolution species under the experimental conditions prevailing in the current study (pH>13, T=80 ��C) (Eq. (4)).
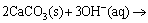
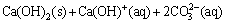 (4)
(4)
Kinetic investigations on calcite dissolution have shown that the reaction kinetics of calcite is significantly slower than the diffusion kinetics, which tends to make it reaction-limited, so, temperature could be an effective parameter [29-31]. The rate of calcite dissolution was found to be relatively independent of pH between 8 and 10 [28] while it decreases by about an order of magnitude over the pH range of 10-14 [25]. In the case of dolomite, results showed that both calcite and dolomite undergo congruent dissolution while dolomite dissolves ~5 times slower than calcite [29]. Dissolution rates of dolomite were found to be pH-independent at 6��pH��8 and to decrease with increasing pH at pH>8. Dissolution of dolomite results in Ca2+ and Mg2+ in addition to ions. At elevated temperature (>80 ��C) Ca2+ and Mg2+ will precipitate in the form of CaCO3 and Mg(OH)2 at pH >12.5, respectively [32,33]. Accordingly, dolomite dissolution reaction under alkaline condition can be described as
 (5)
(5)
To sum up, it can be concluded that the dissolution of calcite and dolomite is expected to happen at elevated pH and temperature such as that applied in this study and this process can affect pyrite oxidation which will be discussed in the following parts.
3 Experimental
3.1 Samples
A natural pyrite sample was collected from a rock sample of the Mazraeh Copper Deposit, Ahar, Iran, where sulfide mineralization is extensive. The large crystals of pyrite were carefully removed from the rock. The pyrite sample was crushed and physically fractionized to +37-53 ��m. The treated sample was identified as cubic pyrite by X-ray diffractometry (Fig. 1) with minor amount of chalcopyrite as the main gangue mineral. Pyrite sample was found to comprise 85% pyrite according to XRF analysis (Table 1). Prior to the leaching experiments, small samples of the ground material were soaked in 3 mol/L hot hydrochloric acid solution for 15 min, filtered, rinsed with distilled water, dried, and kept under vacuum in a desiccator. The acid solution dissolved oxides and other soluble compounds already presented in the raw material or oxides formed during grinding. High-purity (>99%) natural calcite and dolomite were used in this study. All other chemicals were used in analytical grades.
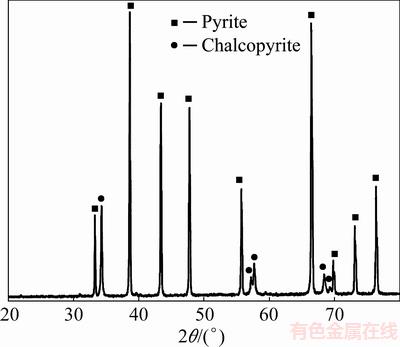
Fig. 1 XRD pattern of pyrite sample
Table 1 XRF analysis of pyrite sample (mass fraction, %)

3.2 Leaching tests
The leaching experiments were performed in a batch way in an Erlenmeyer flask, which was putted on a magnetic stirring hot plate with temperature controlling system. The tests were carried out with 5% solids (5 g pyrite + 5 g gangue mineral per 200 mL solution) and the oxygen flow rate was maintained at 200 cm3/min. Applying such proportion of pyrite and gangue minerals, a saturated solution of calcite or dolomite can be made. Temperature, agitation speed, and pH were controlled throughout the tests; when necessary, pH was adjusted with drops of 1 mol sodium hydroxide solution. The samples were withdrawn periodically, filtered, and the filtrates were submitted to the analytical laboratory for sulfate analysis.
3.3 Solution analysis
The oxidation of pyrite was determined by analyzing the soluble sulfur species produced during the oxidation of pyrite using turbidimetric method [34] in which sulfate ion was precipitated in an acetic acid medium with barium chloride so as to form barium sulfate crystals of uniform size. Light absorbance of the barium sulfate suspension was measured by Unico UV-Vis spectrophotometer and the sulfate concentration was determined by comparison of the data with a standard curve. Since preliminary tests indicated the presence of less oxidized sulfur species, the samples were treated by hydrogen peroxide and also were kept at least for a week in order to include the oxidation of all soluble sulfur species to sulfate. No concentration changes were observed after passing this period of time. The concentration of carbonate ion was determined by direct acid-base titration method using phenolphthalein and methyl orange as indicator [34].
3.4 Determination of initial rate
The conversion of pyrite (X') was calculated using the measured sulfate concentration as follows:
X��=(m0-mt)/m0 (6)
where mt represents the measured sulfate concentration of solution at time t and, m0 represents the total sulfate concentration that can be released into solution after complete oxidation of pyrite. Initial reaction rates were determined by fitting the experimental conversion (X��) data to a second-order polynomial regression equation:
X��=a+bt+ct2 (7)
The coefficient b represents the value of the initial rate (vi) that is given by the derivative of conversion (X') with respect to time:
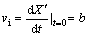 (8)
(8)
3.5 Instrumental analysis
XRD patterns were collected at room temperature and 2��=25��-75��, with a step size of 2��=0.02�� and a collection time of 1 s/step using a Bruker Axs D8 advanced instrument. The solution potential was measured using a silver chloride reference electrode. Potentials are shown relative to the standard hydrogen electrode. Fourier transformed infrared (FTIR) spectra of the samples were recorded in the spectral range of 400-4000 cm-1 using Bruker model instrument via KBr pelletization method. The morphology and composition of solid particles were examined by field emission scanning electron microscopy (FE-SEM) using a TESCAN FE-SEM equipped with energy-dispersive X-ray spectroscopy (EDS).
4 Results and discussion
4.1 Leaching experiments
The increase in the rate of pyrite oxidation with increasing pH, temperature and oxygen overpressure is well established [35,36], the oxidation experiments in this study were conducted at temperature of 80 ��C, initial pHi=13.33 (c(NaOH)=0.4 mol/L) (other values of pH were mentioned) and oxygen overpressure of 1.013��105 Pa. The tests were also done at rotating speeds greater than 500 r/min, so the effect of mass transfer in bulk solution was minimized. Figure 2 shows the effect of calcite and dolomite on the conversion of pyrite where pyrite oxidation yield decreases as calcite or dolomite is added to system.
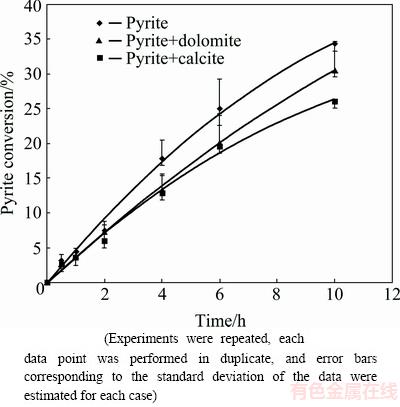
Fig. 2 Effect of calcite and dolomite addition on pyrite oxidation in sodium hydroxide solution at pHi=13.33, T=80 ��C and p(O2)=1.013��105 Pa
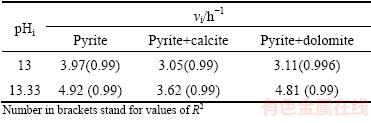
Variations of initial pyrite oxidation rate (vi) with pH are demonstrated in Table 2. Detrimental effect of both calcite and dolomite on pyrite oxidation rate can be seen at pH>13 although dolomite has less negative effect on pyrite oxidation than calcite.
Table 2 Effect of pH on initial rate (vi) of pyrite oxidation
The reduction in pyrite oxidation yield in the presence of carbonate minerals can be attributed to the variation in system parameters such as pH or solution potential as a result of calcite or dolomite dissolution. The formation of passive insoluble layer on pyrite surface is another possible reason. pH variation during pyrite oxidation is illustrated in Fig. 3. According to Fig. 3, solution pH decreases by passing reaction time which is due to OH- ions consumption during pyrite oxidation according to Eq. (1). pH reduction order (pH(pyrite)��pH(pyrite+dolomite)��pH(pyrite+calcite)) is in agreement with pyrite oxidation rates (Fig. 2), showing that higher oxidation rates result in the lower values of the solution pH.
It is interesting to note that there is an irregular variation in pH at initial stage of the reaction where more pH reduction during pyrite oxidation is observed in the presence of calcite and dolomite than that in the absence of these minerals. This abnormal behavior can be attributed to OH- ions consumption due to the dissolution of carbonate minerals according to Eqs. (4) and (5), which leads to a decrease in pH of solution. This idea is supported by the results demonstrated in Fig. 4 showing the concentration of carbonate ions during pyrite oxidation. According to Fig. 4, calcite and dolomite were partially dissolved in alkaline solution up to 15.27% and 15.6% (mass fraction) respectively after 10 h. This results in the formation of ions and other species such as Ca(OH)2 and Mg(OH)2 according to thermodynamic modeling and solid characterization studies provided in the following parts.
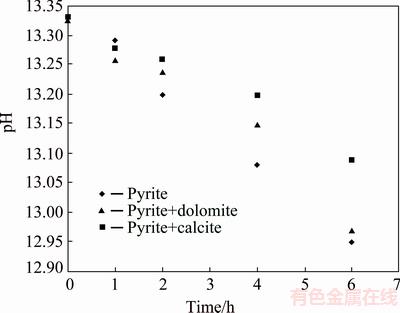
Fig. 3 pH variation during pyrite oxidation at pHi=13.33, T=80 ��C and p(O2)=1.013��105 Pa
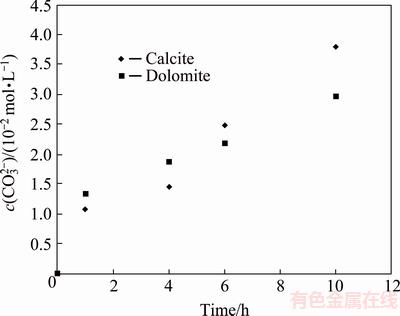
Fig. 4 Carbonate ion concentration during pyrite oxidation in the presence of calcite and dolomite at pHi=13.33, T=80 ��C and p(O2) =1.013��105 Pa
Figure 4 also shows a reduction in carbonate ion concentration in the case of dolomite at final time of the leaching test. This implies the precipitation of carbonate ions from the solution which will be discussed in the chemical modeling section.
Solution potential variation during pyrite oxidation is demonstrated in Fig. 5. The dissolution of carbonates refers to non-oxidative reactions in which the mineral is neither oxidized nor reduced [37], consequently, no significant variation in solution potential is expected due to the presence of calcite and dolomite. Figure 5 shows that the solution potential increases slightly in the presence of carbonate minerals where the pyrite oxidation rate is lower. It can be concluded that increasing solution potential does not significantly affect the oxidation rate of pyrite under alkaline condition. Considering that the higher solution potential results in the accelerated reaction rate in chemical controlled reaction, it can be predicted that the kinetics of pyrite oxidation in the presence of calcite and dolomite does not control chemically and other phenomenon such as mass transfer may affect pyrite oxidation in the presence of carbonate minerals. Therefore, further investigation seems necessary in this area.
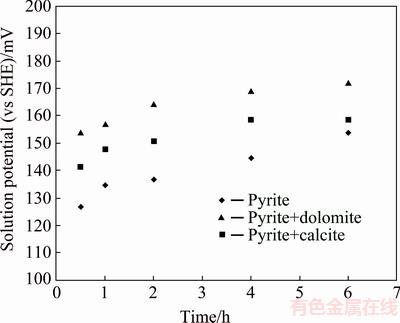
Fig. 5 Solution potential variation during pyrite oxidation at pHi=13.33, T=80 ��C and p(O2) =1.013��105 Pa
Activation energy (Ea) data (Fig. 6) are also consistent with the results under the condition that mass transfer controls the rate of pyrite oxidation. In other words, the addition of carbonate minerals into solution should decrease Ea. The data in Fig. 6 show that upon addition of calcite and dolomite, Ea decreases from 27.66 to 24.73 and 23.52 kJ/mol, respectively. The difference in Ea is small but meaningful. However, the Ea values reported represent apparent activation energies, which include diffusion-controlled reactions on the surface of pyrite as well as adsorption and precipitation. It seems that solid surface characterization is necessary to get a clearer understanding of pyrite oxidation mechanism in the presence of gangue minerals. This is provided in the following parts through chemical modeling, XRD, FTIR and FE-SEM analysis.
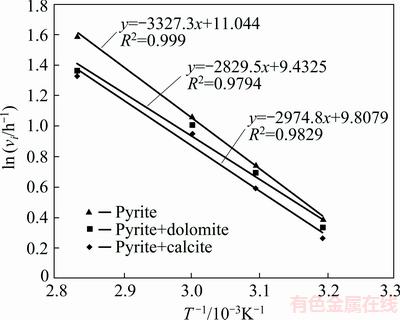
Fig. 6 Activation energy of pyrite oxidation reaction in the presence of calcite and dolomite at pHi=13.33 and p(O2)=1.013��105 Pa
4.2 Chemical modeling
Chemical modeling involving multiple chemical equilibria can be used to predict an aqueous processing system with the required temperature, pH and concentration [32]. Therefore, in this study, the dissolution behavior of calcite and dolomite in NaOH-H2O solutions was investigated and interpreted by chemical modeling plotted with OLISystems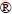 software. Concentrations were chosen based on the experimental results of this study in which about 15% (mass fraction) of calcite and dolomite dissolved during reaction time of 10 h (Fig. 4). The solid lines represent the resulting chemical modeling as a function of pH value at various reaction temperatures ranging from 25 to 80 ��C. Accordingly, chemical modeling was done for two distinct solutions, one containing 0.04 mol/L dissolved calcite and the other with 0.02 mol/L dissolved dolomite. The results shown in Fig. 7 demonstrate the effect of pH and temperature on the dissolution behavior of calcite (Figs. 7(a)-(c)) and dolomite (Figs. 7(d)-(i)). It can be seen from Figs. 7(a) and (d) that the dissolution of calcite and dolomite is favorable under both acidic and alkaline conditions. The curves of calcite and dolomite were shifted to the left with increasing temperature. These results indicate that it is possible to dissolve calcite and dolomite at a much lower pH, rather than that at 25 ��C, by increasing the reaction temperature from 25 to 80 ��C. So, it can be predicted that at 80 ��C calcite and dolomite are no longer thermodynamically stable above pH of 13 and 11, respectively. This prediction is in consistent with the results of leaching experiments concerning partial dissolution of calcite and dolomite under initial pH of 13.33 and at 80 ��C (Fig. 4).
software. Concentrations were chosen based on the experimental results of this study in which about 15% (mass fraction) of calcite and dolomite dissolved during reaction time of 10 h (Fig. 4). The solid lines represent the resulting chemical modeling as a function of pH value at various reaction temperatures ranging from 25 to 80 ��C. Accordingly, chemical modeling was done for two distinct solutions, one containing 0.04 mol/L dissolved calcite and the other with 0.02 mol/L dissolved dolomite. The results shown in Fig. 7 demonstrate the effect of pH and temperature on the dissolution behavior of calcite (Figs. 7(a)-(c)) and dolomite (Figs. 7(d)-(i)). It can be seen from Figs. 7(a) and (d) that the dissolution of calcite and dolomite is favorable under both acidic and alkaline conditions. The curves of calcite and dolomite were shifted to the left with increasing temperature. These results indicate that it is possible to dissolve calcite and dolomite at a much lower pH, rather than that at 25 ��C, by increasing the reaction temperature from 25 to 80 ��C. So, it can be predicted that at 80 ��C calcite and dolomite are no longer thermodynamically stable above pH of 13 and 11, respectively. This prediction is in consistent with the results of leaching experiments concerning partial dissolution of calcite and dolomite under initial pH of 13.33 and at 80 ��C (Fig. 4).
Under alkaline condition the dissolution of calcite and dolomite results in the precipitation of Ca(OH)2 and Mg(OH)2 according to Eqs. (4) and (5). Formation of Ca(OH)+ and Mg(OH)+ is also possible. It can be found from Figs. 7(b) and (c) that Ca(OH)2 is the main product of calcite dissolution while the contribution of Ca(OH)+ becomes more and more negligible as the temperature and base concentration increase. On the other hand, the dissolution behavior of dolomite is different to some extent. While Mg(OH)2 is favorable Mg species under pH of 13 at 80 ��C (Figs. 7(h) and (i)), but Ca2+ precipitates mainly in the form of CaCO3 than Ca(OH)2 under the similar condition of temperature and pH (Figs. 7(f) and (g)). Accordingly, Ca(OH)2 only precipitates at the higher values of pH (pH>13.5) if calcite does not exist in the suspension (Fig. 7(e)) as it is described in Section 2.
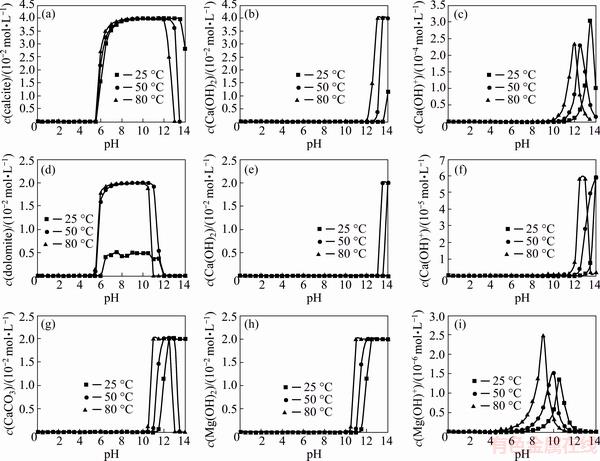
Fig. 7 Chemical modeling of effect of reaction temperature on dissolution and precipitation behavior of calcite (a), Ca(OH)2 (b), Ca(OH)+ (c) in CaCl2-Na2CO3-NaOH-H2O solutions (0.04 mol/L initial amount of CaCl2 and Na2CO3 per alkali solution) and dolomite (d), Ca(OH)2 (e), Ca(OH)+ (f), CaCO3 (g), Mg(OH)2 (h) and Mg(OH)+ (i) in CaCl2-MgCl2-Na2CO3-NaOH-H2O solutions (0.02 mol/L initial amount of CaCl2 and MgCl2 and 0.04 mol/L initial amount of Na2CO3 per alkali solution) at different pH values
4.3 XRD analysis
XRD patterns of solid phases after alkaline leaching of pyrite in the absence and presence of calcite and dolomite are illustrated in Fig. 8. Reactants are detected through relative intense peaks indicating that pyrite oxidation and calcite and dolomite dissolution were incomplete. A weak diffraction peak at 2��=31.01�� can be observed in all patterns, which can be assigned to the diffraction features of goethite (ICDD No. 00-002- 0281). Formation of very poor crystalline iron oxides or oxyhydroxides was reported as oxidation product of pyrite leaching under similar condition of pH and temperature [38,39]. A characteristic peak of Ca(OH)2 was detected at 2��=39.6�� (ICDD No. 01-084-1274) for pyrite oxidation in the presence of calcite showing the precipitation of Ca(OH)2 in this system. It is interesting to note that in the case of dolomite not only the peak of Mg(OH)2 (2��=60.08��, ICDD No. 01-002-1092) but also a diffraction peak of calcite was observed. This is in line with chemical modeling results where it was predicted that in the presence of Mg2+, Ca2+ precipitates mainly in the form of calcite at pH of 13 and 80 ��C.
Fig. 8 XRD patterns of solid products from oxidation of pyrite in the absence and presence of calcite and dolomite at T=80 ��C, pHi=13.33, p(O2)=1.013��105 Pa and reaction time of 10 h
4.4 FTIR analysis
FTIR spectra of pure calcite and dolomite were compared with those of pyrite oxidation in the presence of calcite and dolomite in Figs. 9 and 10, respectively. Tests were done at 80 ��C and pH of 13.33. Samples were dried at 80 ��C for 1 h before conducting analyses. The peaks around 3850 and 3740 cm-1 are assigned to S��S stretching vibration of pyrite. The broad band centered about 3400 cm-1 and the band at 1640 cm-1 were observed in the presence of both calcite and dolomite. These bands are assigned to O��H stretching and bending modes of water or Fe oxyhydroxides (such as hematite and goethite), respectively [40]. Whereas calcite and dolomite dissolution mechanism over the pH range of 8-14 involves the adsorption of water onto the mineral surface [25], so OH bands of water are probable at FTIR spectra. The results of pyrite oxidation in the presence of calcite show bands at 1425-1430, 874, 710 and 2864 cm-1 which are assigned to the ��3, ��2, ��4 and 2��3 stretching vibration of C��O bond at calcite [41,42]. Decreasing intensity of calcite characteristic peaks with pH (Fig. 9) confirms calcite partial dissolution which may result in aqueous and solid Ca(OH)2 based on thermodynamic considerations. Accordingly, the presence of several broad and weak bands at around 3640 cm-1 can be assigned to O��H stretching vibration in Ca(OH)2 [43].
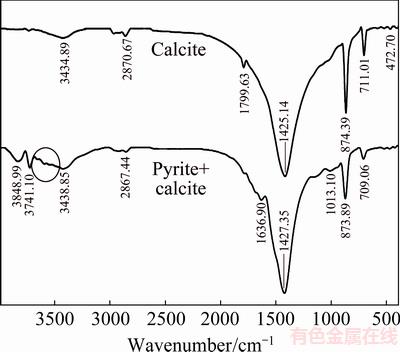
Fig. 9 FTIR spectra of oxidized pyrite in the presence of calcite at T=80 ��C, pHi=13.33, p(O2)=1.013��105 Pa and reaction time of 10 h
FTIR spectra of pyrite oxidation in the presence of dolomite at pH 13 are shown in Fig. 10. The bands at 1445, 878 and 720 cm-1 are assigned to the ��3, ��2 and ��4 stretching vibration of C��O bond of dolomite, respectively. It is observed that band of dolomite at 1445 cm-1 was shifted to 1455 cm-1 and a new band at 1516 cm-1 was developed at pHi 13.33. The extent of splitting (number of distinguishable bands) of the 1455 cm-1 band, representing the free ion, could be directly related to the bond strength between and a metal cation or surface [41]. Thus, it appears to correspond to the formation of calcite as it is predicted through chemical modeling and XRD analysis. Accordingly, the bands at 1455 and 1516 cm-1 can be assigned to the ��3 stretching vibration of C��O bond in dolomite and calcite, respectively. These positions are shifted to longer wavelength transmittance which is in agreement with previous studies where it was reported that band positions of pure calcite and dolomited are places slightly to longer wavelengths as they mixed [44-46]. The presence of several weak bands around 3600 cm-1 confirms new O��H stretching vibration which is attributed to Mg(OH)2.
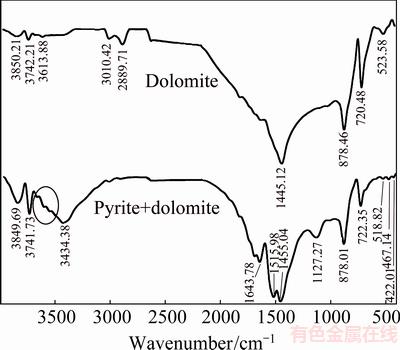
Fig. 10 FTIR spectra of oxidized pyrite in the presence of dolomite at T=80 ��C, pHi=13.33, p(O2)=1.013��105 Pa and reaction time of 10 h
4.5 SEM-EDS analysis
The dissolution of calcite and dolomite was observed by scanning electron microscopy. Figure 11 shows the FE-SEM images of these minerals after 10 h alkaline dissolution at various magnifications. As shown in these images, both calcite and dolomite partially were dissolved under alkaline condition, which results in products with totally different morphologies and textures. There are needle-like nanoparticles which gradually change into agglomerated fine particles in the case of calcite (Figs. 11(a)-(c)). EDS analysis shows that Ca content increases during calcite dissolution and precipitation of new products which support the idea of Ca(OH)2 formation. Discrete morphology was observed in the case of dolomite dissolution products in comparison to that of calcite. It was previously reported that the Mg content of the amorphous calcium carbonate phase directly affected the final morphology of the precipitates [47,48]. It is clear from Figs. 11(d)-(f) that dolomite dissolution forms plate-shape Mg(OH)2 nanoparticles which are agglomerated in porous structure. Similar morphologies have been observed during synthesis of Mg(OH)2 nanoparticles [49,50].

Fig. 11 FE-SEM images concerning calcite (a-c) and dolomite (d-f) dissolution at T=80 ��C, initial pHi=13.33, p(O2)=1.013��105 Pa and reaction time of 10 h

Fig. 12 FE-SEM images of pyrite surface after alkaline leaching in the presence of calcite (a-c) and dolomite (d-f) at T=80 ��C, pHi=13.33, p(O2)=1.013��105 Pa and reaction time of 10 h
Pyrite surface morphologies after alkaline oxidation in the presence of calcite and dolomite are demonstrated in Fig. 12 at various magnifications. Formation of a surface layer is obvious in the presence of both calcite and dolomite. Figure 12 shows that the external surface layer is thin and almost continuous in the presence of calcite while it seems thick and discontinuous in the case of dolomite which grows gradually on the surface of pyrite and finally breaks into solution. The above results illustrate the distinct morphology of the oxide coatings formed during pyrite oxidation in NaOH solution in the presence of different gangue minerals, under the same experiment conditions. This is in line with chemical modeling prediction and also with solid characterization studies discussed in previous parts in which Ca(OH)2 was proposed as the main product of calcite dissolution while calcite and Mg(OH)2 were detected during dolomite dissolving. Among them Ca(OH)2 shows higher tendency to adsorb on pyrite surface. This can be related to the precipitation mechanism of Ca(OH)2 on pyrite surface through the formation of Ca(OH)+. It was reported that the adsorption of Ca(OH)+ on pyrite surfaces was stronger than the adsorption of OH- which results in the formation of Ca(OH)2 on pyrite surface [51,52]. On the other hand, this cannot be true about Mg(OH)2 because Mg(OH)+ is not stable at pH>11 (Fig. 7(i)). These results fortify the idea that the formation of an external layer slows down the further oxidation reaction and also justifies more detrimental effect of calcite than dolomite on pyrite oxidation rate.
5 Conclusions
1) Pyrite alkaline oxidation rate decreases in the presence of calcite and dolomite. Activation energies for pyrite dissolution are calculated to be 24.73 and 23.52 kJ/mol in the presence of calcite and dolomite, respectively, which implies that the dissolution is controlled by mass transfer.
2) Characterization study of the residue confirmed that calcite and dolomite were partially dissolved during alkaline oxidation. The reaction products were identified to be Ca(OH)2 and Mg(OH)2.
3) Morphology analyses showed that products of calcite and dolomite dissolution cover pyrite surface during alkaline leaching. It was also found that surface layer is thin and continuous in the case of calcite while it is thick and discontinuous in the presence of dolomite.
4) Based on the above discussion, it was suggested that the presence of calcite and dolomite slows down pyrite oxidation due to precipitation of their alkaline dissolution products on pyrite surface.
5) Chemical modeling data showed that calcite and dolomite solubility is affected dominantly by the pH and temperature of the leaching solution. So, careful control of solution parameters is necessary to prevent the formation of passive layer on pyrite surface. The application of dispersants and additives may also be beneficial which need further investigation.
Acknowledgments
The authors acknowledge the support of IMIDRO, Iranian Mines & Mining Industries Development & Renovation, for providing the financial support to carry out this research.
References
[1] MURPHY R, STRONGIN D R. Surface reactivity of pyrite and related sulfides [J]. Surface Science Reports, 2009, 64: 1-45.
[2] CHANDRA A P, GERSON A R. The mechanisms of pyrite oxidation and leaching: A fundamental perspective [J]. Surface Science Reports, 2010, 65: 293-315.
[3] AGORHOM E A, SKINNER W, ZANIN M. Influence of gold mineralogy on its flotation recovery in a porphyry copper�Cgold ore [J]. Chemical Engineering Science, 2013, 99: 127-138.
[4] GUO X Y, XIN Y T, WANG H, TIAN Q H. Mineralogical characterization and pretreatment for antimony extraction by ozone of antimony-bearing refractory gold concentrates [J]. Transactions of Nonferrous Metals Society of China, 2017, 27: 1888-1895.
[5] YANNOPOULOS J C. The extractive metallurgy of gold [M]. Heidelberg: Springer, 1991.
[6] MARCHEVSKY N, BARROSO QUIROGA M M, GIAVENO A, DONATI E. Microbial oxidation of refractory gold sulfide concentrate by a native consortium [J]. Transactions of Nonferrous Metals Society of China, 2017, 27: 1143-1149.
[7] MELASHVILI M, FLEMING C, DYMOV I, MATTHEWS D, DREISINGER D. Dissolution of gold during pyrite oxidation reaction [J]. Minerals Engineering, 2016, 87: 2-9.
[8] MELASHVILI M, FLEMING C, DYMOV I, MATTHEWS D, DREISINGER D. Equation for thiosulphate yield during pyrite oxidation [J]. Minerals Engineering, 2015, 74: 105-111.
[9] YANG Y B, ZHANG X, XU B, LI Q, JIANG T, WANG Y X. Effect of arsenopyrite on thiosulfate leaching of gold [J]. Transactions of Nonferrous Metals Society of China, 2015, 25: 3454-3460.
[10] CELEP O,  H, VICIL M. Characterization of refractory behaviour of complex gold/silver ore by diagnostic leaching [J]. Transactions of Nonferrous Metals Society of China, 2009, 19: 707-713.
H, VICIL M. Characterization of refractory behaviour of complex gold/silver ore by diagnostic leaching [J]. Transactions of Nonferrous Metals Society of China, 2009, 19: 707-713.
[11] CELEP O, SERBEST V. Characterization of an iron oxy/hydroxide (gossan type) bearing refractory gold and silver ore by diagnostic leaching [J]. Transactions of Nonferrous Metals Society of China, 2015, 25: 1286-1297.
[12] ASADI H H, VONCKEN J H L, HALE M. Invisible gold at Zarshuran, Iran [J]. Economic Geology, 1999, 94: 1367-1374.
[13] MEHRABI B, YARDLEY B W D, CANN J R. Sediment-hosted disseminated gold mineralisation at Zarshuran, NW Iran [J]. Mineralium Deposita, 1999, 34: 673-696.
[14] ASADI H H, VONCKEN J H L, K��HNEL R A, HALE M. Petrography, mineralogy and geochemistry of the Zarshuran Carlin-like gold deposit, northwest Iran [J]. Mineralium Deposita, 2000, 35: 656-671.
[15] MESA ESPITIA S L, LAPIDUS G T. Pretreatment of a refractory arsenopyritic gold ore using hydroxyl ion [J]. Hydrometallurgy, 2015, 153: 106-113.
[16]  D, THIBAULT Y. Influence of potassium hydroxide pretreatment on the extraction of gold and silver from a refractory ore [J]. Hydrometallurgy, 2014, 146: 64-71.
D, THIBAULT Y. Influence of potassium hydroxide pretreatment on the extraction of gold and silver from a refractory ore [J]. Hydrometallurgy, 2014, 146: 64-71.
[17] KOSLIDES T, CIMINELLI V S T. Pressure oxidation of arsenopyrite and pyrite in alkaline solutions [J]. Hydrometallurgy, 1992, 30: 87-106.
[18] LI W, LIU Z, HUANG Q, TANG Y, QIU X. Extraction of low-grade silver from a refractory Au-Ag ore in cyanidation by pretreatment with reductive alkaline leaching [J]. Hydrometallurgy, 2016, 164: 257-264.
[19] ZHONG S P. Leaching kinetics of gold bearing pyrite in H2SO4-Fe2(SO4)3 system [J]. Transactions of Nonferrous Metals Society of China, 2015, 25: 3461-3466.
[20] CALDEIRA C L, CIMINELLI V S T, DIAS A, OSSEO-ASARE K. Pyrite oxidation in alkaline solutions: Nature of the product layer [J]. International Journal of Mineral Processing, 2003, 72: 373-386.
[21] CALDEIRA C L, CIMINELLI V S T, OSSEO-ASARE K. The role of carbonate ions in pyrite oxidation in aqueous systems [J]. Geochimica et Cosmochimica Acta, 2010, 74: 1777-1789.
[22] SENANAYAKE G. Role of copper(II), carbonate and sulphite in gold leaching and thiosulphate degradation by oxygenated alkaline non-ammoniacal solutions [J]. Minerals Engineering, 2005, 18: 409-426.
[23]  LG, MARTELL A E, BJERRUM J. Stability constants of metal-ion complexes [M]. London: Chemical Society, 1964.
LG, MARTELL A E, BJERRUM J. Stability constants of metal-ion complexes [M]. London: Chemical Society, 1964.
[24] SHERMAN L A, BARAK P. Solubility and dissolution kinetics of dolomite in Ca-Mg-HCO-3/CO32- solutions at 25 ��C and 0.1 MPa carbon dioxide [J]. Soil Science Society of America Journal, 2000, 64: 1959-1968.
[25] FREDD C N, SCOTT FOGLER H. The kinetics of calcite dissolution in acetic acid solutions [J]. Chemical Engineering Science, 1998, 53: 3863-3874.
[26] REARDON E J, FAGAN R. The calcite/portlandite phase boundary: Enhanced calcite solubility at high pH [J]. Applied Geochemistry, 2000, 15: 327-335.
[27] REGNAULT O, LAGNEAU V, SCHNEIDER H. Experimental measurement of portlandite carbonation kinetics with supercritical CO2 [J]. Chemical Geology, 2009, 265: 113-121.
[28]  E L. A fundamental equation for calcite dissolution kinetics [J]. Geochimica et Cosmochimica Acta, 1976, 40: 441-447.
E L. A fundamental equation for calcite dissolution kinetics [J]. Geochimica et Cosmochimica Acta, 1976, 40: 441-447.
[29] YADAV S K, CHAKRAPANI G J, GUPTA M K. An experimental study of dissolution kinetics of calcite, dolomite, leucogranite and gneiss in buffered solutions at temperature 25 and 5 ��C [J]. Environmental Geology, 2008, 53: 1683-7694.
[30] COLOMBANI J. The alkaline dissolution rate of calcite [J]. The Journal of Physical Chemistry Letters, 2016, 7: 2376-2380.
[31] HEYDARPOUR T, REZAI B, GHARABAGHI M. A kinetics study of the leaching of a calcareous phosphate rock by lactic acid [J]. Chemical Engineering Research and Design, 2011, 89: 2153-2158.
[32] UM N, HIRATO T. Precipitation behavior of Ca(OH)2, Mg(OH)2, and Mn(OH)2 from CaCl2, MgCl2, and MnCl2 in NaOH-H2O solutions and study of lithium recovery from seawater via two-stage precipitation process [J]. Hydrometallurgy, 2014, 146: 142-148.
[33] HU Z, DENG Y. Synthesis of needle-like aragonite from calcium chloride and sparingly soluble magnesium carbonate [J]. Powder Technology, 2004, 140: 10-16.
[34] CLESCERI L S. Standard methods: For the examination of water and wastewater [M]. Washington: American Public Health Ass, 1998.
[35] CIMINELLI V S T, OSSEO-ASARE K. Kinetics of pyrite oxidation in sodium hydroxide solutions [J]. Metallurgical and Materials Transactions B, 1995, 26: 677-685.
[36] CIMINELLI V S T, OSSEO-ASARE K. Kinetics of pyrite oxidation in sodium carbonate solutions [J]. Metallurgical and Materials Transactions B, 1995, 26: 209-218.
[37] CRUNDWELL F K. The mechanism of dissolution of minerals in acidic and alkaline solutions: Part I��A new theory of non-oxidation dissolution [J]. Hydrometallurgy, 2014, 149: 252-264.
[38] CUDENNEC Y, LECERF A. The transformation of ferrihydrite into goethite or hematite, revisited [J]. Journal of Solid State Chemistry, 2006, 179: 716-722.
[39] KUKKADAPU R K, ZACHARA J M, FREDRICKSON J K, SMITH S C, DOHNALKOVA A C, RUSSELL C K. Transformation of 2-line ferrihydrite to 6-line ferrihydrite under oxic and anoxic conditions [J]. American Mineralogist, 2003, 88: 1903-1914.
[40] MAZZETTI L, THISTLETHWAITE P J. Raman spectra and thermal transformations of ferrihydrite and schwertmannite [J]. Journal of Raman Spectroscopy, 2002, 33: 104-111.
[41] NAKAMOTO K. Applications in inorganic chemistry: Infrared and Raman spectra of inorganic and coordination compounds [M]. New York: John Wiley & Sons, Inc, 2008.
[42] GUNASEKARAN S, ANBALAGAN G, PANDI S. Raman and infrared spectra of carbonates of calcite structure [J]. Journal of Raman Spectroscopy, 2006, 37: 892-899.
[43] LEGODI M A, de WAAL D, POTGIETER J H, POTGIETER S S. Rapid determination of CaCO3 in mixtures utilising FT-IR spectroscopy [J]. Minerals Engineering, 2001, 14: 1107-1111.
[44] ZAINI N, van der MEER F, van der WERFF H. Effect of grain size and mineral mixing on carbonate absorption features in the SWIR and TIR wavelength regions [J]. Remote Sensing, 2012, 4: 987-1003.
[45] van der MEER F. Spectral reflectance of carbonate mineral mixtures and bidirectional reflectance theory: Quantitative analysis techniques for application in remote sensing [J]. Remote Sensing Reviews, 1995, 1: 67-94.
[46] GAFFEY S J. Spectral reflectance of carbonate minerals in the visible and near infrared (0.35-2.55 um): Anhydrous carbonate minerals [J]. Journal of Geophysical Research: Solid Earth, 1987, 92: 1429-1440.
[47] LOSTE E, WILSON R M, SESHADRI R, MELDRUM F C. The role of magnesium in stabilising amorphous calcium carbonate and controlling calcite morphologies [J]. Journal of Crystal Growth, 2003, 254: 206-218.
[48] SANTOS R M, CEULEMANS P, van GERVEN T. Synthesis of pure aragonite by sonochemical mineral carbonation [J]. Chemical Engineering Research and Design, 2012, 90: 715-725.
[49] PARVEEN M F, UMAPATHY S, DHANALAKSHMI V, ANBARASAN R. Synthesis and characterization of nanosized Mg(OH)2 and its nanocomposite with poly (vinyl alcohol) [J]. Nano, 2009, 4: 147-156.
[50] HANLON J M, DIAZ L B, BALDUCCI G, STOBBS B A, BIELEWSKI M, CHUNG P. Rapid surfactant-free synthesis of Mg(OH)2 nanoplates and pseudomorphic dehydration to MgO [J]. Cryst Eng Comm, 2015, 17: 5672-5679.
[51] LI Y, CHEN J, KANG D, GUO J. Depression of pyrite in alkaline medium and its subsequent activation by copper [J]. Minerals Engineering, 2012, 26: 64-69.
[52] HUANG H, HU Y, SUN W. Activation flotation and mechanism of lime-depressed pyrite with oxalic acid [J]. International Journal of Mining Science and Technology, 2012, 22: 63-67.
Ehsan BIDARI, Valeh AGHAZADEH
Department of Mineral Processing, Faculty of Mining Engineering, Sahand University of Technology, Tabriz, Iran
ժ Ҫ��ͨ����Ъ�������飬��ϻ�ѧģ�⡢��Һ������������������о�����ʯ�Ͱ���ʯ�Ի�����������Ӱ�졣��80 ��C��p(O2)=1.013��105 Pa��pH>13�������½��н������顣����������ڷ���ʯ�Ͱ���ʯ���������£��������ڼ��е��������½������з���ʯ��Ӱ�����FE-SEM/EDS������������������溬�и�(����ʯ����ʱ)��þ(����ʯ����ʱ)�����ǻ�����������½���ԭ���ݻ�ѧģ��������һ�������γ�������̼���ο���IJ����ܽ⣬��Ҫ�ܽ���ҺpHֵ���¶ȵ�Ӱ�졣XRD��FTIR���������������Ϊ�ƻ�þ����������о������֣��ڷ���ʯ���ڵ�����£�����㱡�������������ģ����ڰ���ʯ���������£������Ⱥ��ֲ�������
�ؼ��ʣ����������̼������ʯ�������ͽ��
(Edited by Wei-ping CHEN)
Corresponding author: Valeh AGHAZADEH; Tel: +98-4133459235; Fax: +98-4133444311; E-mail: v.aghazadeh@sut.ac.ir
DOI: 10.1016/S1003-6326(18)64782-X

