�й���ɫ����ѧ�� 2004,(11),1850-1855 DOI:10.19476/j.ysxb.1004.0609.2004.11.011
���������ױ�Ĥ���������ƻ��ķ��Ӷ���ѧģ��
�Ƶ� ��ΰ�� ����ľ
�㽭��ѧ��ѧϵ,�㽭��ѧ��ѧϵ,�㽭��ѧ��ѧϵ ����310027 ,����310027 ,����310027
ժ Ҫ��
Ӧ�÷��Ӷ���ѧ����ģ���˵��������ױ�Ĥ�ܵ��������ƻ��Ĺ���,�ó����׳߶ȵ�������Ĥ��Ӧ����Ӧ���ϵ�������ݻ����ߺ�����Ĥ���͵ı仯�����˵��γɺ���չ���̡�ģ�����ԭ����Ƕ������ԭ�Ӽ�����,�õ���������Ĥ�ĵ���ģ��,���������������ϵͳԭ��������Ӧ���仯������صĹ�ϵ���������:���ױ�Ĥ�����ɱ���Ӱ�����������ԭ�ӵ��˶��ͱ�Ĥ������ѧ����,���ױ�Ĥ�ƻ��ļ���������ԭ�ӿ�λ�����Ӻ;���ȱ�ݵ���չ;�����Ķ��ѽӽ����Զ���,ģ��õ����ױ�Ĥ�Ķ���ǿ�ȷ���Griffith���Զ��ѵ�����ƽ�����ۡ�
�ؼ��ʣ�
���Ӷ���ѧ ;�� ;�������� ;��Ƕԭ�ӷ� ;
��ͼ����ţ� TB383
����飺 �Ƶ�(1979),��,��ʿ�о���.ͨѶ����:�ơ���;�绰:0571 87951769;E mail:huang524@zju.edu.cn;
�ո����ڣ� 2004-03-27
���� ������Ȼ��ѧ�������������Ŀ(10302025);
Molecular dynamics simulation for failure process of monocrystalline nickel film under tensile stress
Abstract��
The failure process of monocrystalline nickel film under tensile stress was simulated with molecular dynamics method. The evolvement of atomic energy and arrangement of atoms in the model, the initiation and expansion of damage and the elastic modulus of monocrystalline nickel were obtained. Simulation results show that the free surfaces of single crystal take effect on the motion of atoms and mechanical properties of nano crystal. Atomic cavities and the growth of crack in crystal lead to the failure of nano film. The fracture of single crystal is similar to macro brittle rupture, and the fracture strength of the film can be explained well by Griffith's theory of rupture.
Keyword��
molecular dynamics; nickel; tension; embedded atom method;
Received�� 2004-03-27
�������ϵͳ(MEMS)�о�������������ϵͳ(NEMS)�ķ�չ, �ɹ۳߶��²�����ѧ�� �������ܵ��о��Ե��������С� ����ʵ����Ʒ�Ʊ���ʵ��������Ƶ�����, Ŀǰ��������ѧʵ��ͨ�������ڶԲ��ϵ���ģ���� Ӳ�ȡ� ���Ȳ����IJ���
[1 ,2 ,3 ]
, ��ʵ�����������ϴ�, ��ʹ�����ģ���Ϊ�о��ɹ۳߶��²�����Ϊ����Ҫ����֮һ��
���Ӷ���ѧ����(MD)��Ŀǰ�ɹ۳߶�ģ�����Ҫ�ֶΡ� ��ֱ�Ӹ���ԭ�Ӽ�������ģ��ԭ�ӵ��˶�����, �ҳ�������ṹ����ѧ���ܵı�����ϵ�� ��ԭ�Ӽ�������ƺ�����ѡȡ�Ƿ��Ӷ���ѧ���㾫ȷ���Ĺؼ��� ԭ����Ƕ��(EAM)
[4 ]
�����˴�ͳ�Ķ���ģ�ͶԶ���ЧӦ���Dz���ֵ�ȱ��, ���Ը���ȷ���������ڽ��桢 ���桢 ȱ�ݵȲ�������ԭ���˶���
����������Ͻ��������¡� ����ʴ�� �������õĻ�ѧ�ȶ���, ��ҽ����е�� ������ ������ ��������������õ��㷺Ӧ�á� ���в����������������Ļ�ѧ�������ʡ� �������ϲ��ϵĺ������
[5 ,6 ,7 ]
, ���������ɹ۳߶�����ѧ�� ��е���ܵ�����
[8 ]
��Խ��١�
��������Ӧ�÷��Ӷ���ѧ�����о����������ܵ������������ƻ��Ĺ��̡� ����ԭ����Ƕ������ԭ�Ӽ�����ý����˵�������Ĥ����ά����ģ��, ͨ��ԭ��ģ�⿼���˱�Ĥԭ�������� Ӧ���ı仯�ͽṹ���ι��̡� ���ױ�Ĥȱ�ݵIJ����� �����Լ���չ����, �õ����ױ�Ĥ����̬���������µĵ���ģ���� Ӧ����Ӧ���ϵ�� �����ݻ�����, ���������ɱ��桢 λ���˶���ԭ�ӻ��ƶ����ײ������ܺͱ��ε�Ӱ�졣 ��������ƽ��Ƕ���Griffith�������۽����˵�������Ĥ�Ķ��ѡ�
1 ���Ӷ���ѧģ��
1.1 ģ��ԭ���뷽��
��Ƕԭ�ӷ������ڽ�����ѧ����ģ�⡣ ���о�����Voter��
[9 ]
������Ƕԭ�ӷ���������Ķ����ƺ����� ��ԭ��������Ϊ
E
t
o
t
a
l
=
��
i
[
1
2
��
j
��
(
r
i
j
)
+
F
(
��
i
)
]
?
?
?
(
1
)
ʽ�� �� (r ij r ij i , j ֮������Ķ���; F (�� i �� i �� i i ���ĵ������ܶȡ� ����
�� (r ij A 1 (r c 1 r ij 2 exp(-c 1 r ij
F
(
��
i
)
=
D
��
i
ln
��
i
��
i
=
��
j
f
(
r
i
j
)
?
?
?
(
3
)
����
f (r ij A 2 (r c 2 r ij 2 exp(-c 2 r ij
ʽ(4)��ʾ��ԭ��i ����Ϊr ij j ��ԭ��i �������Ƶ�Ӱ�졣 A 1 �� A 2 �� c 1 �� c 2 �� D Ϊ�ɲ����������ܾ����IJ����� r c 1 r c 2 r ij
[
10 ]
������������������Ƕ�ƺ��������ķ���, ȡr c 1 d , �ڴν���ԭ�Ӻ͵�������ԭ��֮��, r c 2 d , �ڵ������ں͵��Ľ���ԭ��֮�䡣 d Ϊ�����ԭ�Ӿ���, ����������������,
d
=
a
0
/
2
?
a
0
��������
ԭ�Ӽ����������ţ�ٵڶ��˶�����
F
i
=
-
?
E
t
o
t
a
l
?
r
i
=
m
i
?
v
i
?
?
?
(
5
)
ʽ�� F i r i m i v i
[11 ]
���ٶ���ʽ
[12 ]
������֡�
1.2 ģ�͵Ľ���
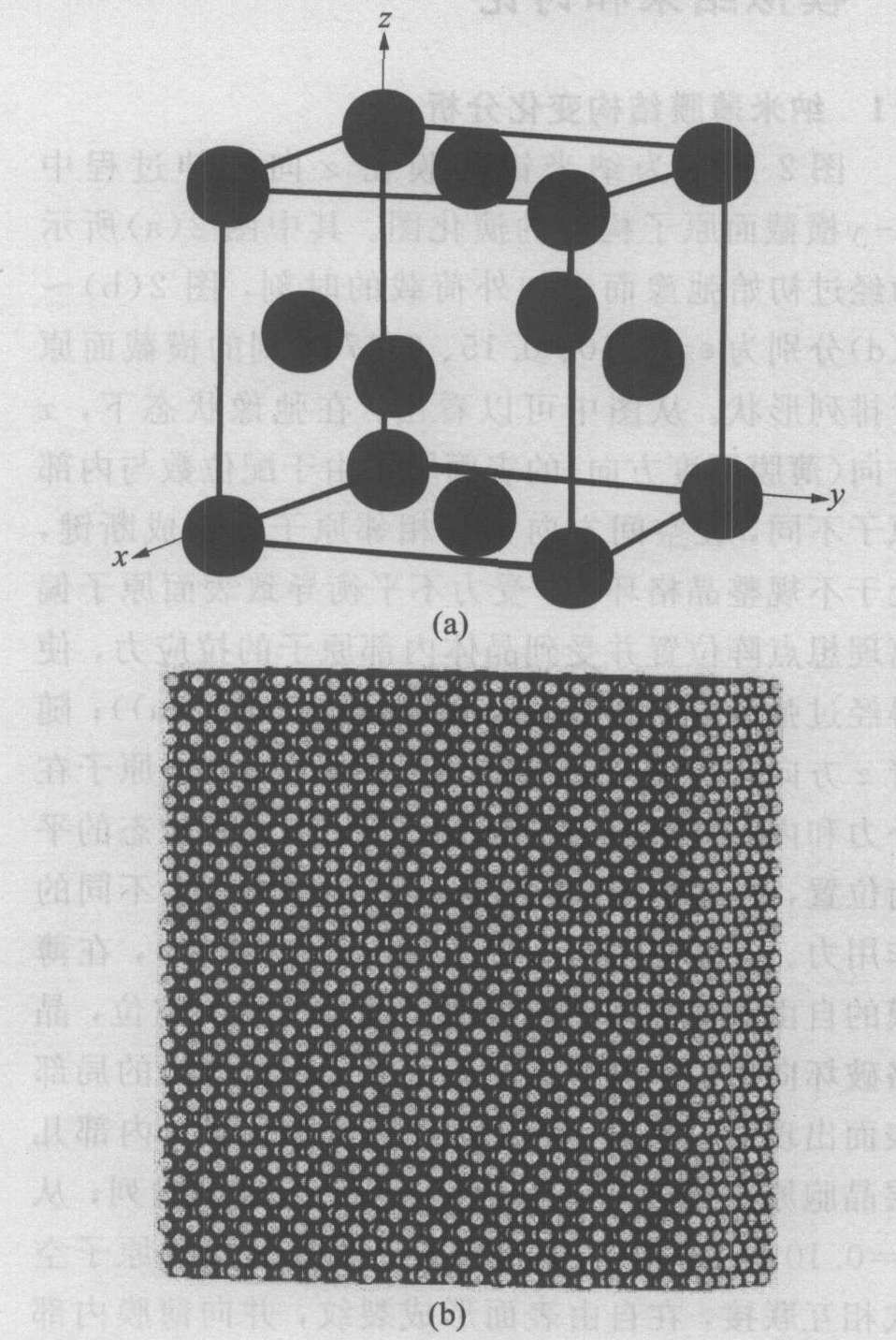
ͼ1��ʾΪ��������Ĥԭ��ģ��ʾ��ͼ�� ͼ1(a)��ʾΪ�����������ṹ(fcc)����ʾ��ͼ, ͼ1(b)Ϊ����ģ�͡� x , y , z ������ֱ��Ӧ������[100], [010], [001]���� ���ù���ߴ�, Ϊʹ�ɹ۱�Ĥ�Ӿ�������, ȡ6��20��20������, ��9 600��ԭ�ӡ� ģ��ԭʼ�ߴ�Ϊ2.112 nm��7.04 nm��7.04 nm�� ģ���г��ȵ�λΪ���ľ�����a 0 =0.352 nm, ����ϵͳ�������ȶ�����, ���Ӷ���ѧģ���ʱ�䲽����Ϊt 0 =3.5��10-15 s, ������λΪE 0 =10-19 J�� ��x ����ı߽����Ϊ���ɱ���, ��y , z ������ʩ�������Ա߽�����, ʹԭ��ģ�ͳʵ�����Ĥ�ṹ�� ��ϵͳ�¶ȳ�ʼ��Ϊ�������, ����ģ���б�������, ����ԭ���ȼ�� ��ʵ������ԭ�ӵ����˶����ı��������ģ��������ѧ�����仯��������ơ�
ͼ1 ��������Ĥԭ��ģ��
Fig.1 Atomic model of nano nickel film
(a)��Nickel lattice; (b)��Simulation model
1.3 ģ�����
��ģ�ͽ����¶ȳ�ʼ��, �������¶�ʼ��Ϊ�������, ����ԭ���ȼ�� ���ȶԵ���ģ��ԭ�ӳ�ԥ20 000��, ʹϵͳ�г��ʱ��ﵽ������͵��ȶ�״̬�� ��z ����ʩ��ƽ������Ӧ��0.005, ���з��Ӷ���ѧģ�����10 000��, ʱ��Ϊ3.5��10-11 s, Ȼ���ԥ20 000��, ʹϵͳ�ص�ƽ��̬�� ����������Ӧ��0.005, �ظ���ʩ��Ӧ�䡪���Ӷ���ѧģ�⡪��ԥ����, ʹģ��ԭ�Ӵ�����̬��������״̬�� ��������в���Parrinello-Rahman
[13 ]
��������x , y ����ijߴ��������������ѹ������, ������һ������ѹ�� ������̬����ģ������Ĥ�г���ԭ�ӿ�λʱ, ����ʩ�Ӻ�������, ��Ӧ��������Ϊ0.001, �ظ���������, ���Ĥ�ֲ��ƻ�,�ƻ����ͱ�Ĥ�������ڴ���ԭ�Ӱ���ϵͳ�����ö��뿪ģ��ռ�, ģ������� ģ�������ÿ��100����¼ԭ�ӵ�Ӧ��ʸ��, ������, ����, ����, ��Ĥ��״�� ������������״��ԭ������λ�á�
2 ģ����������
2.1 ���ױ�Ĥ�ṹ�仯����
ͼ2��ʾΪ��������Ĥ��z �����������x -y �����ԭ�ӹ��͵��ݻ�ͼ�� ����ͼ2(a)��ʾΪ������ʼ��ԥ��δ������ص�ʱ��, ͼ2(b)��2(d)�ֱ�Ϊ�� =0.10�� 0.15�� 0.17ʱ�̵ĺ����ԭ��������״�� ��ͼ�п��Կ���: �ڳ�ԥ״̬��, x ����(��Ĥ��ȷ���)�ı���ԭ��������λ�����ڲ�ԭ�Ӳ�ͬ, �ڿռ䷽��ʧȥ����ԭ�Ӷ��γɶϼ�, ���ڲ����������� ������ƽ��±���ԭ��ƫ���������λ�ò��ܵ������ڲ�ԭ�ӵ���Ӧ��, ʹ�þ�����ԥ��ı�Ĥ��������������(ͼ2(a)); ����z ��������Ӧ�������, ���渽���ļ���ԭ�����������ڲ�ԭ�������������¿�ʼƫ���ԥ̬��ƽ��λ��, ������λ�õı仯���ڲ�ԭ�Ӳ�����ͬ���������� ��Ӧ��ﵽ0.10ʱ(ͼ2(b)��ʾ), �ڱ�Ĥ�����ɱ���������Ե�ԭ���˶����µĿ�λ, �����ƻ���Ĥ�ڲ����뼸��ԭ��, �ڽ��䴦�ľֲ���������غ����45�㷽��չ�Ļ����档 �ڲ����㾧��ԭ�����л�������, ��Ϊ�����������; ���� =0.10���� =0.15�Ĺ�����, ��Ĥ�����ԭ�ӿ�Ѩ�����, �����ɱ����γ�����, ����Ĥ�ڲ���չ(ͼ2(c)), �ڲ������ƻ��������ӡ� ���ڼ�������Ϊ��̬, �������Ƽ���������㹻��ʱ��������λ��, ʹ�Ѽ�ۻ�, �ʲ����ڴ˽α��ֳ���������; ����ؼ�������ʱ, ��Ĥ������������, ������������������, ���д�������ԭ��������ȫ�ͷ�, ���뱡Ĥϵͳԭ�ӵ�����, �뿪ģ��ռ�, ��Ĥ�ڲ�������λ���ɹۿ������� ���ӵ��±�Ĥ�����췽���ϳ��ֹᴩ��Ĥ��ȵ���������, ��Ĥ��y ��Ҳ���ڶ���(��ͼ2(d)), ģ�������
ͼ2 ���ױ�Ĥx-y�����ԭ������
Fig.2 Atomic arrangement on x -y section in nano nickel film
2.2 �����ݻ����̷���
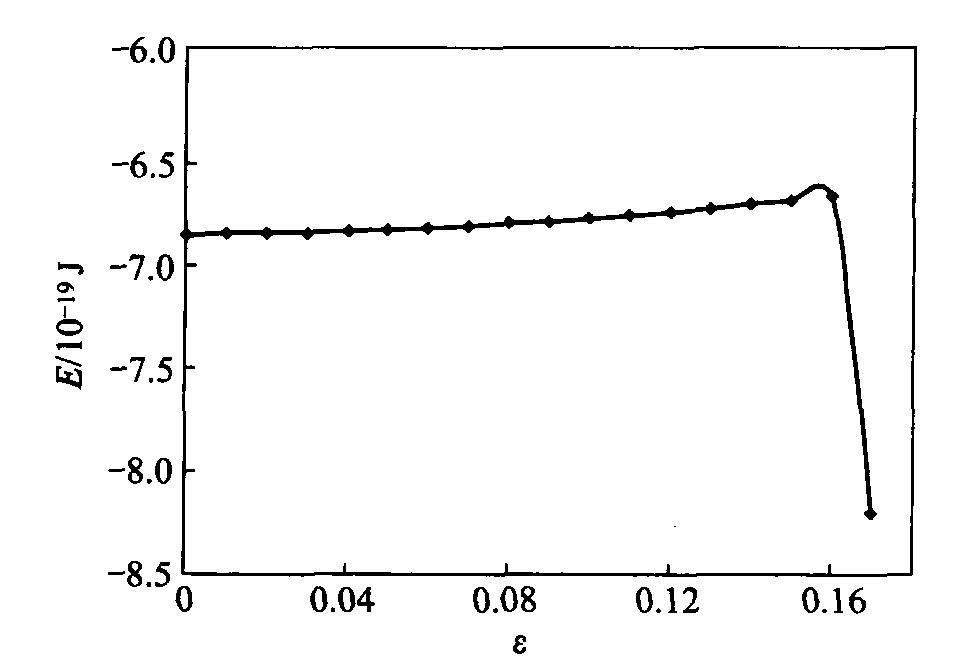
ͼ3��ʾΪ���������ԭ��ƽ����������Ӧ��仯����ֵ������ߡ� �ڱ��о����� =0.03��ǰȡģ��ȫ��ԭ����������ƽ��ֵ, ���˺����ģ�͵ı����ص�, ȡͼ2�к����λ�ø�����10��ԭ��������ƽ���� ��ͼ��֪, �ӳ�ʼ��ԥ����״̬����������ӵ��� =0.03��ʱ��, ϵͳ��������ά����һ��ˮƽ����, ������Ϊ���ױ�Ĥ�ȱ������, ����ԭ�����ڶԳ��Ա�����, ԭ����λ����, �ڿռ䷽���γɶϼ�, ���ڳ�ʼ��ԥ�δ����˴����ı����ܡ� �ڳ�ʼ����ʱ, �ڲ�ԭ��������������˶�, ��������, ����Ҳ��ƽ��̬��ʼ����, ������Ĥ�ڲ�ԭ������������, ������ԭ�������������¿�ʼ�ͷ����ʼ������ƽ�������Ĵ���������, ��ʹϵͳ��������ά�ֻ�������; ���� =0.03��ʼ, ϵͳ��������ʼ��������, �˺����ű���ԭ�ӱ����ܵ���ȫ�ͷ�, ��������Ĥϵͳԭ����������������������ߡ� �˺�, ԭ�ӻ��Ըߵı��澧�����ȿ�ʼ�����ɹ��ƻ�����, ����ԭ���������������ƫ������λ�ø���, ����ԭ�ӿ�λ�Ϳ��� ԭ���˶��Ӿ�, ϵͳ���������ٶ�Ҳ����(���� =0.08���� =0.14)�� ����, �ڴ˽��оֲ��ı���ԭ����һ��������, ������λ���ƶ�Ҳ���IJ���ԭ�������� ���� =0.14���� =0.16�Ĺ�����, ģ���ƻ�����ϵͳԭ�����л���, ��Ĥ��ȱ�ݵ��������չʹ����ԭ���˶�Զ��ƽ��λ��, ԭ��ƽ��������������, �ɹ�ȱ�ݵ����ӳ������ƻ����������ɶȸߵ�ԭ��֮����볬���ضϰ뾶, �γ��µı���, ģ�������췽�����, �±�����γ�ʹ�����㲿�ֵ�ԭ�����������������ֵ(ͼ3); �˺��������췽��ȡһ�����ּ�������, �ɼ��ڱ�Ĥ��ȷ���Ҳ����ֹᴩ����������, ��Ĥ��y �������ڶ��ѡ� ���������췽������Ժ�, ģ���еĴ���ԭ���뿪ģ��ռ�, ϵͳ��������Ѹ���½���
ͼ3 ����Ĥԭ����������Ӧ��ı仯
Fig.3 Variation of atomic total energy with stress in nickel film
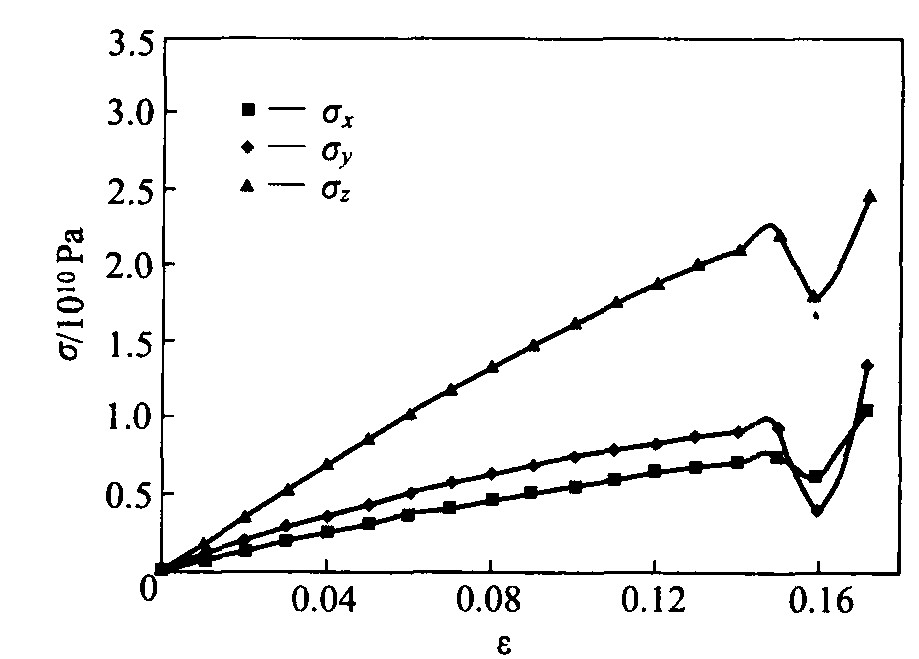
2.3 ԭ��Ӧ����Ӧ�����
Ӧ����Ӧ���ϵ�ڲ��ϵ������о����к���Ҫ�����á� ͼ4��ʾΪ����Ĥ����������ù����е�Ӧ���ݻ����ߺ������췽���Ӧ����Ӧ���ϵ, ������Ӧ��Ϊϵͳ��ԭ�ӵ�ƽ����Ӧ���� ���ڱ�Ĥ�ں�ȷ��������������, ���ڱ�Ĥƽ���־��ж�ά�Գ���, ���z �����Ӧ����Ӧ���ϵ�͵���ģ�����Ӧͬ��������y ���� ��ģ���ʼʱ��(�� =0), �������ɳ�ԥ������ģ���ڲ�ԭ��Ӧ��Ϊ��, �����ɱ���ԭ�����ڿռ��ϲ��Գ�, �ܵ��ڲ�ԭ�ӵ�����, ���漸��ԭ��֮��������������ϵͳԭ��ƽ��Ӧ���������㡣 ��ģ���������֪, �ھ�����ԥ���������״̬, �� x 4 Pa, �� y �� z 5 Pa, ������ԥ״̬����ԭ���ر�Ĥƽ�������Ӧ��������ȷ����ϵ�ԭ���������� ��ͼ4���Կ���, �����������, �����췽�����Ӧ���� z �� y �� x �� =0.06��0.14��, ���ڱ�Ĥ���ɱ���ֲ����ƺ��ڻ�����ĩ�˲���������ȫλ��, ���ڲ���������ȱ��ʱ, ���ɹ�ȱ�ݸ���(���ɹ����Ƶļ��)Ҳ����λ��, λ�����ƶ������������, Ӧ�����ߵ�б�����½�, ʹ���ױ�Ĥ���ֳ��������ԡ� ���ɹ۱�ĤӦ����Ӧ�����߲�û�г��ֺ�۲�������ʵ�������Ե�������, ��֤�˴�ǰ���Ӷ���ѧģ���о�����ϻ���������Ӧ��ǿ���Ľ���
[14 ]
�� �ھ���ԭ�ӻ��ƽκ�, ��������ص�����, ���� =0.148ʱ��, ���ƻ����������췽��ֱ����ļ��㾧����ȱ������, ������������, ��Ĥ������z�������, �����������Ӧ�����ﵽ��ֵ�� �˺�������ԭ�ӳ�ԥ��ȡ����ģ�ͼ�������, ���� =0.17ʱ�غ�ȷ���Ҳ���ֹᴩ����(ͼ2(d)), ��Ĥ������y ����Ҳ���ѡ�
ͼ4 ����Ĥԭ�ӵ�Ӧ����Ӧ������
Fig.4 �� ���� curves of atoms in nickel film
�ӳ�ʼ���ص����ѵ�Ӧ����Ӧ�����߱������ױ�Ĥ�����췽���Ӧ���仯��, �ұ仯���������ԡ� �����غ�ȷ���ͱ�Ĥƽ���ڵĻ��Ƽ�����λ���Ĵ���, �����췽��ֱ����������Ӧ���仯�ϻ����� �������������췽���Ӧ����Ӧ�����������ں�۴��Բ���, ���췽���Ӧ�����ֵΪ�� z
2.4 ����ģ�����
�ɼ��ع����е�Ӧ���� Ӧ��ֵ, ���Ծ�����С���˷��������
[15 ]
�õ����ײ��ϵĵ���ģ���� ��Ӧ�����Ա仯��(�� =0��0.01, ��ǰ�ķ�����֪�˽α�Ĥ���ɱ���ԭ�����ͷű�����, �ڲ�������Ϊ��������, ԭ����ƽ��״̬�����˶�, ��Ĥ����δ�������ñ��λ��γ��ɹ�ȱ��), ���غ������½�Ϊ0.001, ȡģ�������������췽���Ӧ���� Ӧ��ֵ�������, �õ�����ģ��Ϊ186.6 GPa�� ��ֵС�ڶྦྷ���ĵ���ģ��(204.00��220.64 GPa), ҲС��Shen
[16 ]
�ȶ�����϶������������Ʒ�IJ��Խ��(217 GPa)�� ��Ϊģ���в��õ����ױ�Ĥģ���д����ı���ԭ���˶����ɶȸ�, ԭ�Ӽ����ǿ���½�, �����˲��ϵ�ǿ�ȡ� ֵ��ע�����, ���о�������õĵ���ģ����Ϊ��������Ĥ��100������ĵ���ģ����
2.5 �����ݻ��������ƻ�����
��ͼ2������Ĥ�ƻ���չ�������ԭ�����еı仯�ͱ�����������Ӧ�����ݻ����̷�����֪���ױ�Ĥģ���в������˲����� �ݻ����ƻ����̡� ��ʼ��ԥʱ, Ϊ�ﵽ�ȶ�״̬, ��Ĥ���ɱ�������⼸��ԭ���ܵ�����, ƫ���������λ��, ʹ��Ĥ���������״, �������˴���������; ���ܵ���Ĥ���ڵ�������Ӧ������ʱ, �ڲ�����ԭ�����������������ƽ��λ�ø�����, �����Դ�ı���ԭ�����������������ͷű�����, ���澧�������״̬�ɳ�; ��������ص�����, ���ɱ���ֲ�ԭ�ӿ�ʼ�뿪����λ��, ����ԭ�ӿ�λ�� ���ֱ��澧��ԭ����һ��������, ���ڻ������ն˳���������ȫλ��; �����ƻ��ӱ������ڲ�����, ���ڱ�Ĥ����ֲ��������ԵĻ�����(ͼ2(b))�� ��Ӧ��ϴ�ʱ, ȱ��(���ɹ����Ƽ��)������������λ���ڱ�Ĥƽ��ͺ�ȷ�����ƶ������ⲿ����, �����ƺ�λ�����¾����ƻ�������ȱ������ӳ���, �ڱ�Ĥ�ڲ�������������, �������Ƽ����չ�� ����ԭ�ӿ�λ�������λ�����䵼�±�Ĥ�غ������÷���(z ����)�������ƺ�۵ľ�������(ͼ2(c))�� �����λ�ͱ�Ĥ�ڲ����Ƶ���չʹ��Ĥ��y ���췽����ѡ� z ������Ѻ�ı�Ĥ����ԥ��ѡȡ���ּ���ʩ�������, ����ԭ������ϵͳ�������ݳ���Ĥ����, ��Ĥ��y ����(��ֱ�ں��ط���)��Ҳ�������ӡ� ��չ���ᴩ��Ĥ��ȵ���������, ʹ��Ĥ�������ڶ���(ͼ2(d))�� ���ױ�Ĥ�Ķ��Ѳ�û�����Ե�������, ��������������ӵĹ��������ڽ������Ƶ���������ͻȻ����, ˵����Ĥ���Բ���, ��������������ྦྷ���ϲ�ͬ�ı��λ��ƾ����ġ�
����Griffith����������Բ�������ƽ��ľ���������ۼ��������ڷ�������Բ��ϵ���������, ���뾧�����ʱ�����������ϵͳӦ�����ܺ�����Ѳ��������ɱ�����Ӧ�ﵽƽ��, ������ѵ����Ӧ��Ӧ���ھ���ṹ���Գ��ܵ����Ӧ��������ǿ�ȡ� �ڱ��о���ģ���б�Ĥ�����췽�����ʱ, ԭ�������Ӧ���� z
[
16 ]
����������ѧ���ܵIJ���, ����ģ��E =217 GPa, ��ģ�������Ķ���ǿ���Ѿ��dz��ӽ����������ǿ��(һ��ȡΪ0.1E ), ��ȫ����Griffith�����뾧������ƽ��������ۡ�
3 ����
�Ե��������ױ�Ĥ�ھ�̬���������ƻ����̵ķ��Ӷ���ѧģ�����: ���ױ�Ĥ���ڱȱ������, ����������Ըߡ� �����ߵ�����ԭ�ӵĴ���, Ӱ���˲��ϵ������� Ӧ�����Ժͱ��λ��ơ� �����������, ԭ����������������, �����췽�����Ӧ�����, �������췽�����Ȳ�����������, ���±�Ĥ����; �����Ӧ���Сʱ��Ĥ���ɱ���ԭ���ͷű�����, �ڲ�������ԭ��������λ�ø����� Ӧ�����ӵ�һ���̶�ʱ����ԭ���뿪����λ���γɿ�λ�;���ȱ��, ȱ�ݸ�����������λ��, λ�����ƶ�ʹ��Ĥ���ֳ������ԡ� ��������Ӧ����Ӧ������������������, �ӽ���۴��Բ��ϵ������������; ���ɱ���ԭ���˶����¿�Ѩ�����Ӻ��������ڵĻ����Լ�ȱ�ݵ���չ������Ĥ���ѵļ�������; ���ڴ�������ԭ�Ӵ����IJ��ȶ���, ģ�����õ������ڡ�100������ĵ���ģ��E =186.6 GPa, С�ڶྦྷ����ʵ��������������ĵ���ģ��; ģ��õ�����Ĥ����ǿ��Ϊ21.08 GPa, ��������ʵ����������������������ǿ��(ԼΪ21.7 GPa), ����Griffith�������뾧���������ƽ��Ĵ�Ͼ������ۡ�
�����
[1] ��SiegelRW,FouregeGE.Mechanicalpropertiesofnanophasemetals[J].NanostructuredMaterials,1995,6(2):205216.
[2] ��YangMC,YeF,SunXK,etal.Studyonmicrohardnessofbulknanocrystallinecopper[J].NanosturcturedMaterials,1997,9:481484.
[3] ��SandersPG,RittnerM,KiedaischE,etal.CreepofnanocrystallineCu,PdandAl Zr[J].NanostructuredMaterials,1997,9:433440.
[4] ��DawMS,BaskesMI.Embeddedatommethod:Derivationandapplicationtoimpurities,surfacesandotherdefectsinmetals[J].PhysicalReviewB,1984,29(12):64436453.
[5] ������,�졡�|,����ϼ,��.�������Ͻ�Ni/Ni3Al����ķ��Ӷ���ѧģ��[J].����ѧ��,2003,52(10):25202524.WENYu hua,ZHUTao,CAOLi xia,etal.Ni/Ni3AlgrainboundaryofNi basedsinglesuperalloys:Moleculardynamicssimulation[J].ActaPhisicaSinica,2003,52(10):25202524.
[6] ��ZhengYF,CaiW,ZhangJX,etal.High resolutionelectronmicroscopystudyonthesubstructureofTi Ni HfB19'martensite[J].MaterialsLetters,1998,36(3):142147.
[7] ��Rodriguez SantiagoL,TortajadaJ.ExperimentalandTheoreticalstudiesonthegasphasereactivityofformamide Ni+complexesgeneratedbyFABandelectrosprayionization[J].InternationalJournalofMassSpectrometry,2002,219(3):429443.
[8] ��HorstermeyerMF,BaskesMI.Atomisticfinitedeformationsimulation:Adiscussiononlengthscaleeffectsinrelationtomechanicalstresses[J].JournalofEngineeringMaterialsandTechnology,1999,121(2):115119.
[9] ��VoterAF.ChenSP.AccurateinteratomicpotentialsforNi,Al,andNi3Al[A].MaterialsResearchSocietySymposiumProceeding,1987,82:175180.
[10] ��DoyamaM,KogureY.Embeddedatompotentialsinfccandbccmetals[J].ComputationalMaterialsScience,1999,14:8083.
[11] ��AllenMP,TildesleyDJ.ComputerSimulationofLiquids[M].Oxford:ClarendonPress,1987.7882.
[12] ������߮,����ϲ,��㰲,��.����ͭ˿�ߴ�ЧӦ�ķ��Ӷ���ѧģ��[J].��ѧѧ��,2002,34(2):208215.LIANGHai yi,WANGXiu xi,WUHeng an,etal.Moleculardynamicssimulationoflengthscaleeffectsontensionnanocrystalinecopperwire[J].ActaMechanicaSinica,2002,34(2):208215.
[13] ��ParrinelloM,RahmanA.Polymorphictransitionsinsinglecrystals:Anewmoleculardynamicsmethod[J].JournalofAppliedPhysics,1981,52(12):71827190.
[14] ��SchiotzJ,VeggeT,TollaFDD,etal.Atomic scalesimulationsofthemechanicaldeformationofnanocrystallinemetals[J].PhysicalReviewB,1999,60(17):1197111983.
[15] ��ChenZY,YuanQ,DingJQ.Moleculardynamicssimulationsontheconsolidationprocessandrelaxedstructureofnanocrystalline iron[J].ChinesePhysicsLetters,1993,10(2):103106.
[16] ��ShenTD,KochCC,TsuiTY,etal.OntheelasticmoduliofnanocrystallineFe,Cu,NiandCu Nialloyspreparedbymechanicalmilling/alloying[J].JournalofMaterialsResearch,1995,10(11):28922896.