γ-B28的电子结构和弹性性质第一性原理研究
马松山,徐慧,夏庆林
(中南大学 物理科学与技术学院,湖南 长沙,410083)
摘 要:利用第一性原理对硼的高压相γ-B28的能带结构、态密度、分态密度、布居数分布和弹性性质进行研究。研究结果表明:γ-B28具有半导体能带结构的特征,是具有宽间接带隙的半导体材料,且整个带结构由杂化的硼原子2p态和2s态组成,在价带低能段及高能段的-14.5~-11.5 eV区域,硼2p态和2s态对总态密度的贡献大体相当,而在价带高能段的-11.5~0 eV区域及整个导带,对总态密度的贡献主要来源于2p态电子;γ-B28中的化学键具有共价和离子混合键的特征,且共价特征占主导地位;γ-B28单晶弹性常数cij满足力学稳定性判据,表明γ-B28具有力学稳定性,且沿a方向强度最大,而c方向强度最小;γ-B28的体弹模量、剪切模量和弹性模量都较大,而体弹模量和剪切模量的比值BH/GH<1.75及泊松比ν≈0.1,表明γ-B28是一种超硬脆性材料。
关键词:γ-B28;电子结构;弹性性质
中图分类号:TB303 文献标志码:A 文章编号:1672-7207(2010)04-1457-05
First-principle calculation for electronic structure and elastic properties of γ-B28
MA Song-shan, XU Hui, XIA Qing-lin
(School of Physical Science and Technology, Central South University, Changsha 410083, China)
Abstract: First-principles calculations were carried out to investigate the band structure, density of states, partial densities of states Mulliken population and elastic properties of γ-B28 using the method of the local density approximation (LDA) and generalized gradient approximation (GGA) based on density functional theory. The band structure of γ-B28 indicates that it is a kind of semi-conducting material with wide-band gap, and the band structure is hybrid of 2p and 2s electron. The Mulliken population indicates that there is a characteristic of mixed covalent and ionic bonds, and the covalent bond plays a major role. The single-crystal elastic constants cij of γ-B28 obeys the elastic stability criteria, indicating γ-B28 are mechanically stable. In addition, the bulk modulus, shear modulus and elastic modulus of γ-B28 are very large, while BH/GH<1.75 and Poisson’s ratio ν≈0.1, indicating that it is a kind of super-hard and brittle material.
Key words: γ-B28; electronic structure; elastic properties
自从2001年Eremets等[1]发现硼在160 GPa高压下具有超导特性以来,人们对硼进行了大量研究[2-11]。硼的同素异形体较多,但常压下其稳定相到目前仍未在实验室条件下实现。硼的复杂性来源于其在元素周期表中的特殊位置。它处于金属和绝缘体之间,具有3个价电子,倾向于具有金属特性,但是,这些电子又是局域化的,很容易形成绝缘态。当然,这种金属性和绝缘态的微妙平衡关系可以通过压强、温度及掺杂来调控。目前,人们已经知道硼的4个纯相,即α-B12,β-B106,四方T-192和正交γ-B28[3-10],其中γ-B28是最新合成的硼的高压相[6-8]。Oganov等[7]通过实验和理论研究发现γ-B28具有部分离子特性,其离子特性影响γ-B28的禁带宽度、红外吸收和介电常数等性质,并指出γ-B28中化学键以共价键为主。Zarechnaya等[8]通过单晶X线衍射实验和第一性原理计算发现,γ-B28中的化学键本质上为共价键。随后,Godec等[9]研究了γ-B28的状态方程,Rulis等[10]预测了其带边结构的电子能量损耗谱(ELNES),Jiang等[11]研究了其力学性质,揭示γ-B28是一种超硬材料。在此,本文作者利用第一性原理计算方法研究了γ-B28的电子结构和力学特性。首先,通过几何结构优化,得到γ-B28的晶格参数,在此基础上,计算γ-B28的能带结构、态密度、分布密度、化学键的布居数分布及其弹性性质。
1 计算模型和方法
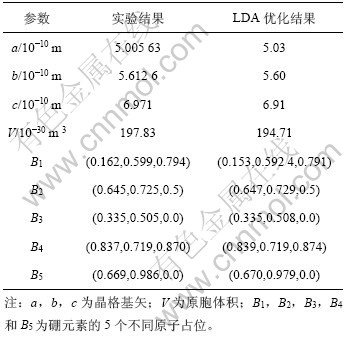
γ-B28属于正交晶系,其晶体结构的空间群为Pnnm(No.58),常压下晶格参数及B元素的5个不同的原子占位如表1所示[6, 8]。计算采用基于密度泛函理论(Density functional theory)结合平面波赝势方法的Castep(Cambridge serial total energy package)第一性原理计算软件包,波函数利用平面波展开[12],交换关联函数采用局域密度近似(LDA)[13]来表示,电子-离子相互作用采用模守恒赝势[14]来表示。对几何结构优化后,晶格参数及原子占位如表1所示。从表1可知:理论计算值与实验结果较吻合。在计算电子结构时,第一布里渊区积分的K点采样利用Monkhorst和Pack格式[15]。截止能量取400 eV,布里渊区K点取样的网格参数选为5×4×4。在弹性常数的计算过程中,交换关联函数分别采用局域密度近似(LDA)及广义梯度近似(GGA)[13, 16]这2种不同方法,选取每一个应变对应的迭代次数为6次,最大应变位移为0.003×10-10 m。
表1 γ-B28的晶格参数
Table 1 Crystal structure data of γ-B28
2 结果和讨论
2.1 γ-B28电子结构
图1所示为γ-B28在布里渊区沿高对称方向的电子能带结构。由图1可知:γ-B28能带价带的最高点和导带的最低点不在同一个K点处,在Г点γ-B28具有价带最大值,在U点具有导带最小值,间接带隙宽约为1.61 eV。可见:γ-B28呈现明显的半导体能带结构的特征,是具有宽间接带隙的半导体材料。
图2所示为γ-B28的态密度(DOS)和分态密度(PDOS)图。由图2可知:整个带结构由杂化的硼原子2p态和2s态组成。同时,对比图1还可看出:γ-B28的价带主要分为2个区,即-16.5~-15 eV的低能段和-14.5~0 eV的高能段。在γ-B28价带低能段及高能段的-14.5~11.5 eV区域,硼2p态和2s态对总态密度贡献大体相当,而在价带高能段的-11.5~0 eV区域及整个导带,对总态密度贡献主要来源于2p态电子,2s态电子贡献较小。
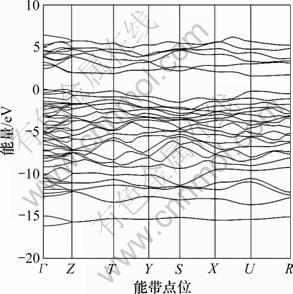
图1 γ-B28的能带结构
Fig.1 Band structure of γ-B28

图2 γ-B28的态密度
Fig.2 Density of states of γ-B28
2.2 γ-B28布居分析
Mulliken布居分析用于分析材料的化学键特性。布居数较高表明键为共价键,布居数较低表明键为离子键,正(负)值表明成键态(反成键态)。表2所示的Mulliken布居分析结果表明:γ-B28中的化学键具有共价和离子混合键的特征,且共价特征占主导地位。这与文献[7-8]中的结论一致。
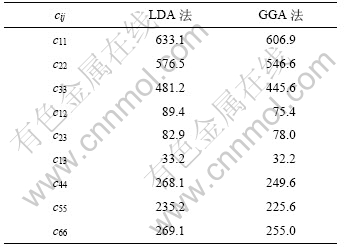
2.3 γ-B28弹性性质
由于γ-B28属于正交晶系,因而有9个相互独立的单晶弹性常数。表3给出了计算得到的弹性常数cij。对于正交晶系而言,其力学稳定性的判据为[17]:
c11>0, c22>0, c33>0, c44>0, c55>0, c66>0,
c11+ c22+ c33+2(c12+ c13+ c23)>0
c11+c22-2c12>0, c11+c33-2c13>0,
c22+c33-2c23>0 (1)
从表3可知:γ-B28的单晶弹性常数cij满足力学稳定性判据,具有力学稳定性。同时,可以发现c11>c22>c33,说明在γ-B28中,沿a方向的强度最大,而c方向的强度最小。
由上述方法计算得到的单晶弹性常数,可进一步计算出γ-B28的弹性模量(体弹模量BH、剪切模量GH、弹性模量E和泊松比ν)。在Voigt近似[18-20]下,γ-B28的体弹模量BV、剪切模量GV可表示为:
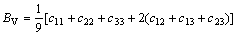 (2)
(2)
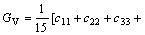
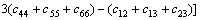 (3)
(3)
表2 γ-B28的Mulliken化学键布居分析
Table 2 Mulliken population analysis of γ-B28
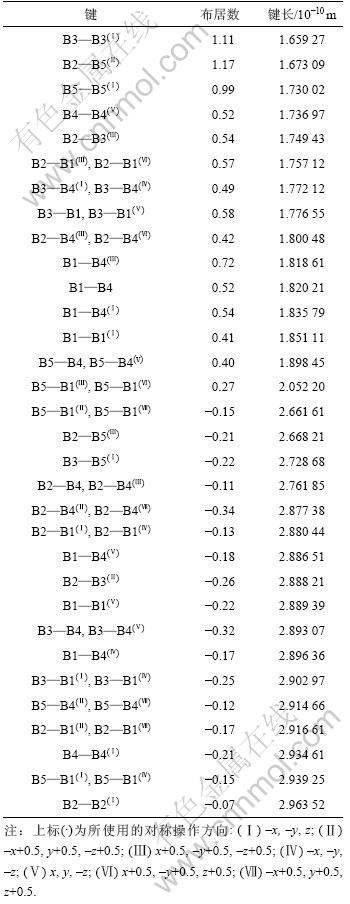
表3 γ-B28的单晶弹性常数cij
Table 3 Single-crystal elastic constants cij of γ-B28 MPa
而在Reuss近似[21]下,其体弹模量BR、剪切模量GR可表示为:
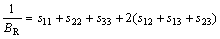 (4)
(4)
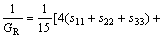
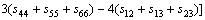 (5)
(5)
其中:sij=1/cij。Connetable等[21]证明了Voigt近似和Reuss近似实际上分别代表了多晶情况下的上限和下限情况,因此,通过对二者的数学平均(Hill近似)可以得到理论上最佳的多晶弹性模量,即:
 ,
, 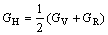 (6)
(6)
同时,依据Hill平均值方法,弹性模量E和泊松比ν可表示为:
 ,
, 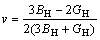 (7)
(7)
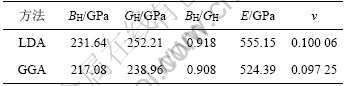
通过计算,可得出γ-B28的体弹模量BH、剪切模量GH、弹性模量E和泊松比ν,见表4。从表4可知:γ-B28的体弹模量BH、剪切模量GH都非常大,表明它是一种超硬材料,并与实验值较吻合[8]。由于BH/GH可以表征物质的韧性和脆性,BH/GH越大,物质的韧性越好,通常BH/GH>1.75代表材料呈韧性,BH/GH<1.75代表材料呈脆性,从表4可知:γ-B28属于脆性材料。同时,弹性模量E和泊松比ν是描述材料弹性行为的2个重要物理量,弹性模量E可以表征材料的刚度,材料的刚度随弹性模量的增大而增强;泊松比ν可以表征固体材料最大拉伸强度与最大剪切强度之比,按照断裂行为的判据,低泊松比的材料属于脆性材料。计算结果显示,γ-B28具有很大的弹性模量,说明它具有很大的刚度,而其泊松比很低(ν≈0.1),表明它是脆性材料,这与BH/GH的计算结果一致。
表4 γ-B28的体弹模量、剪切模量、弹性模量和泊松比
Table 4 Bulk modulus BH, shear modulus GH, elastic modulus E and Poisson’s ratio ν of γ-B28
3 结论
(1) γ-B28具有明显的半导体能带结构的特征,是具有宽间接带隙的半导体材料,且整个带结构由杂化的硼2p态和2s态组成,2p态占主导地位。
(2) γ-B28具有共价和离子混合键的特征,且共价特征占主导地位。
(3) γ-B28单晶弹性常数满足力学稳定性判据,表明γ-B28具有力学稳定性,且沿a方向强度最大,沿c方向强度最小;同时,γ-B28的体弹模量、剪切模量和弹性模量都很大,而体弹模量和剪切模量的比值BH/GH<1.75及泊松比ν≈0.1,表明它是一种超硬脆性材料。
参考文献:
[1] Eremets M I, Struzhkin V V, Hemley R J, et al. Superconductivity in boron[J]. Science, 2001, 293(5528): 272-274.
[2] Sanz D N, Loubeyre P, Mezouar M. Equation of state and pressure induced amorphization of β-boron from X-ray measurements up to 100GPa[J]. Phys Rev Lett, 2002, 89(24): 245501.1-245501.4.
[3] Ma Y Z, Prewitt C T, Zou G T, et al. High-pressure high-temperature X-ray diffraction of β-boron to 30 GPa[J]. Phys Rev B, 2003, 67(17): 174116.1-174116.6.
[4] Masago A, Shirai K and Yoshida H K. Crystal stability of α and β-boron[J]. Phys Rev B, 2006, 73(10): 104102.1-104102.10.
[5] Shirai K, Masago A, Yoshida H K. High-pressure properties and phase diagram of boron[J]. Phys Stat Sol B, 2007, 244(1): 303-308.
[6] Zarechnaya E Y, Dubrovinsky L, Dubrovinskaia N, et al. Synthesis of an orthorhombic high pressure boron phase[J]. Sci Technol Adv Mater, 2008, 9(4): 044209.1-044209.4.
[7] Oganov A R, Chen J H, Gatti C, et al. Ionic high-pressure form of elemental boron[J]. Nature, 2009, 457(7321): 863-867.
[8] Zarechnaya E Y, Dubrovinsky L, Dubrovinskaia N, et al. Superhard semiconducting optically transparent high pressure phase of boron[J]. Phys Rev Letts, 2009, 102(18): 185501.1-185501.4.
[9] Godec Y L, Kurakevych O O, Munsch P, et al. Equation of state of orthorhombic boron γ-B28[J]. Solid State Communications, 2009, 149(33/34): 1356-1358.
[10] Rulis P, Wang L Y, Ching W Y. Prediction of γ-B28 ELNES with comparison to α-B12[J]. Physica Status Solidi-Repid Research Letters, 2009, 3(5): 133-135.
[11] Jiang C, Lin Z J, Zhang J Z, et al. First-principles prediction of mechanical properties of gamma-boron[J]. Appl Phys Letts, 2009, 94(19): 191906.1-191906.3.
[12] Segall M D, Lindan P J D, Probert M J, et al. First-principles simulation: Ideas, illustrations and the CASTEP code[J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[13] Perdew J P, Zunger A. Self-interaction correction to density-functional approximations for many-electron systems[J]. Phys Rev B, 1981, 23(10): 5048-5079.
[14] Zhang X Y, Chen Z W, Zhang S L, et al. Electronic and optical properties of rock-salt aluminum nitride obtained from first principles[J]. J Phys: Condens Matter, 2007, 19(42): 425231.1-425231.5.
[15] Monkhorst H J, Pack J D. Special points for Brillouin-zone integrations[J]. Phys Rev B, 1976, 13(12): 5188-5192.
[16] Ceperley D M, Alder B J. Ground state of the electron gas by a stochastic method[J]. Phys Rev Lett, 1980, 45(7): 566-569.
[17] Sinko G V, Smirnov N A J. Ab initio calculations of elastic constants and thermodynamic properties of bcc, fcc, and hcp Al crystals under pressure[J]. J Phys Condens Matter, 2002, 14(29): 6989-7005.
[18] Wu Z J, Zhao E J, Xiang H P, et al. Crystal structures and elastic properties of superhard IrN2 and IrN3 from first principles[J]. Phys Rev B, 2007, 76(5): 054115.1-054115.5.
[19] Yang J W, Chen X R, Luo F, et al. First-principles calculations for elastic properties of OsB2 under pressure[J]. Physica B, 2009, 404(20): 3608-3613.
[20] Wang H Y, Chen X R, Zhu W J, et al. Structural and elastic properties of MgB2 under high pressure[J]. Phys Rev B, 2005, 72(17): 172502.1-172502.4.
[21] Connetable D, Thomas O. First-principles study of the structural, electronic, vibrational, and elastic properties of orthorhombic NiSi[J]. Phys Rev B, 2009, 79(9): 094101.1-094101.5.
收稿日期:2009-12-15;修回日期:2010-04-26
基金项目:教育部高等学校博士点专项科研基金资助项目(20070533075);湖南省科技计划项目(2009FJ3004)
通信作者:马松山(1971-),男,湖南隆回人,博士,讲师,从事材料性能计算模拟研究;电话:0731-88652708;E-mail: songshan.ma@yahoo.com.cn
(编辑 刘华森)

