��һ��ԭ���о�Pt-Zrϵͳ�л������ ������/��ģ����ԭ����������������
��Դ�ڿ����й���ɫ����ѧ��(Ӣ�İ�)2013���12��
�������ߣ��� ѩ ��Һ� �� Ҷ ������
����ҳ�룺3704 - 3713
�ؼ��ʣ�Pt-Zrϵͳ����ѧ���ܣ��������ܣ���һ��ԭ�����㣻�����ʣ���ģ����ԭ�����
Key words��Pt-Zr system; thermal properties; elastic property; ab initio calculations; formation enthalpy; bulk module; atomic volume
ժ Ҫ��ͨ����һ��ԭ���ļ��㷽�����о�118�ֲ�ͬ�ṹ��Pt-Zr�м仯�����ѡȡ��ص��������ܣ���ṹ�ȶ��ԡ��������������ʡ����Գ����Լ���ģ���Ƚ��м��㡣���ݼ���ó�����������Ϣ������Pt-Zrϵͳ�Ļ�̬�������ߡ�����õ������������ϢΪδ��������ѧ�����ԭ�ӳ߶�ģ���ṩ�������ݡ���ѡȡ��Pt-Zr�������У���������������ع�ϵ����������ԭ���������������أ�����ģ����ԭ������ɸ�������ع�ϵ��
Abstract: 118 kinds of Pt-Zr phases were established and investigated by considering various structures. Then the related physical properties, such as structural stability, lattice constants, formation enthalpies, elastic constants and bulk moduli, are obtained by ab initio calculations. Based on the calculated results of formation enthalpies, the ground-state convex hull is derived for the Pt-Zr system. The calculated physical data would provide a basis for further thermodynamic calculations and atomistic simulations. For these Pt-Zr compounds, it is found there are a positive linear correlation between the formation enthalpies and atomic volumes, and a negative linear correlation between the bulk modules and atomic volumes.
Trans. Nonferrous Met. Soc. China 23(2013) 3704-3713
Xue BAI1, Jia-hao LI1, Ye DAI2, Bai-xin LIU1
1. Advanced Materials Laboratory, School of Materials Science and Engineering, Tsinghua University, Beijing 100084, China;
2. Shanghai Institute of Applied Physics, Chinese Academy of Sciences, Shanghai 200240, China
Received 25 March 2013; accepted 3 September 2013
Abstract: 118 kinds of Pt-Zr phases were established and investigated by considering various structures. Then the related physical properties, such as structural stability, lattice constants, formation enthalpies, elastic constants and bulk moduli, are obtained by ab initio calculations. Based on the calculated results of formation enthalpies, the ground-state convex hull is derived for the Pt-Zr system. The calculated physical data would provide a basis for further thermodynamic calculations and atomistic simulations. For these Pt-Zr compounds, it is found there are a positive linear correlation between the formation enthalpies and atomic volumes, and a negative linear correlation between the bulk modules and atomic volumes.
Key words: Pt-Zr system; thermal properties; elastic property; ab initio calculations; formation enthalpy; bulk module; atomic volume
1 Introduction
Over the last decades, great efforts have been made in the fundamental understanding of both the phase stability and mechanical properties of intermetallic compounds based on the results of quantum�Cmechanical electronic structure calculations [1-4]. Ab initio calculation based upon electronic density functional theory (DFT) has been employed to derive a number of basic physical properties, including formation enthalpies, lattice parameters, elastic constants, and the relative stability of competing phases [5-8]. YANG et al [9] reported a detailed investigation on the electronic structure, chemical bonding behavior, ground-state properties, and optical properties of the whole M-IRMOF-10 (M=Zn, Cd, Be, Mg, Ca, Sr and Ba) series using first-principles methods. As presented by WANG et al [10], ab initio calculations are employed to determine the formation enthalpy of Cu3Au alloys in either ordered or disordered state. Using the plane-wave pseudopotential method based on the density functional theory (DFT) in ABINIT code, LI et al [11] studied the phase stability, electronic structure and mechanical properties of Ti5Al2C3, a recently synthesized Ti-Al-C ternary compound. Previous research has demonstrated that those ab initio methods yield high accuracy in deriving the formation enthalpies and basic mechanical properties for ordered intermetallic compounds in a wide range of systems.
For Zr-based bulk metallic glasses (BMGs), many Zr-light metal compounds have been studied by experiment and calculation. A constructed n-body potential of the Ni-Zr-Al system was applied by ZHAO et al [12], and Monte Carlo simulations were adopted to study the formation and atomic configurations of ternary metallic glasses. Using direct current magnetron sputtering, SHIN et al [13] synthesized the Zr65Cu35 thin film metallic glass, and examined the plasticity-induced nanocrystalline phase transformation by high resolution transmission electron microscopy. However, for Zr-precious metal ones, most experiments focused on the synthesis of metallic glasses, and reports on computer simulations were much less extensive than those with many commercial applications [14,15]. As TAGUE et al [16] reported, Pt-Zr binary thin film composition spreads were deposited at low Zr concentrations using magnetron sputtering and screened for methanol and ethanol electrooxidation activity using a fluorescence assay. Using first-principles local density functional approach, XING et al [17] calculated the ground-state structural phase stabilities and enthalpies of formation of refractory ZrPt and ZrPt3 compounds. Previous studies commonly focused on special properties or phases of the Pt-Zr compounds, and little systematic or basic researches have been reported for the Pt-Zr binary system. Therefore, the Pt-Zr compounds are taken as objects to supply the bank of basic data. On one hand, the calculated results could be a connection of experiments and thermodynamic calculations, since most physical properties at low temperature could hardly be detected directly. On the other hand, ab initio method is a significant application for fitting empirical potentials which are widely utilized in Molecular Dynamics and Monte Carlo simulations.
2 Method and procedure of calculation
The electronic structure problems were identified by the ab initio simulation package of CASTEP (Version 5.5) [18]. The Perdew-Burke-Ernzerhof functional (PBE) functions [19] for the metals supplied with CASTEP and exchange�Ccorrelation functional gradient-corrected (GGA) [20] were adopted without modification. The crystalline phases were first optimized by the BFGS method [21]. The implementation of BFGS scheme in CASTEP involved a Hessian matrix in the mixed space of internal and cell degrees of freedom, so that both lattice parameters and atomic coordinates can be optimized [22]. Elastic constants were evaluated by calculating the stress tensor for a number of distorted phases. The accuracy of the elastic constants depends to a great extent on the accuracy of the SCF setting and also on the level of convergence of geometry optimizations for each distorted structure. These parameters as the energy cut off, maximum displacement, SCF convergence criteria and k�Cpoint set recommended by CASTEP were adopted in calculation, which balanced the efficiency and precision.
In order to verify the relevance of the ab initio method employed, the lattice stabilities, structural properties as well as elastic constants of Zr and Pt metals were calculated. These data are shown in Tables 1 and 2. It is shown that the calculated physical properties are all in agreement with the available experimental data. Therefore, one can conclude that the ab initio method utilized in present study is reasonable and sufficient enough for our required accuracy.
Table 1 Comparison of lattice stabilities (kJ/mol) of Zr and Pt at 0 K calculated by ab initio

Table 2 Lattice constants (a, c), structural energies (Ec), bulk modulus (B0), and elastic constants (Cij) of Zr and Pt obtained from CASTEP and experiments (second rows) [25]

3 Structural and elastic properties of Pt-Zr compounds
In the present study, 9 stable and 109 hypothetical compounds were selected for the Pt-Zr system to study their structural stability and other related basic physical properties, including lattice constants, formation enthalpies, elastic constants as well as bulk modulus. The phases obtained in experiment (already reported by researchers) are simply supposed to be stable phases, and the others constructed are supposed to be meta-stable ones. All the calculated results are listed in Tables 3 and 4. It can be found that both the calculated lattice constants and formation enthalpies of stable phases match well with available experimental data.
Table 3 Comparison of lattice parameters, cell angles (90�� are not been listed) and formation enthalpies ��H of Pt-Zr compounds obtained by ab inito calculations (at 0 K, in upper rows) and available experimental results (at ambient temperature, in lower rows)

to be continued
Continued

to be continued
Continued

Table 4 Calculated elastic constants and bulk modulus of stable and hypothetical crystalline phases in Pt-Zr system

For the equiatomic PtZr compound, a previous study shows that the room-temperature structure is orthorhombic CrB-type, space group Cmcm (a=3.409 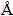 , b=10.315 , c=4.277 ). The orthorhombic structure exits stably up to 1848 K, but then is replaced by a cubic B2-type phase with a=3.3851 at 1863��10 K [27, 34]. In experiment, the orthorhombic phase re-appeared on cooling, indicating that there is a typically martensitic transformation existing in the compound. As presented in Table 3, the calculated results show that the lattice parameters of the orthorhombic CrB-type phase are a=3.445 , b=10.436 , c=4.328 , and that of the B2-type phase is a=3.356 . The deviation of lattice constants is in the order of 0.01 or in the range of 1.17%, indicating that the calculated results are in accordance with the experimental ones. Simultaneously, it is found that the formation enthalpy of the orthorhombic phase is about 20 kJ/mol lower than that of B2-type phase at 0 K, providing a thermodynamic basis for the fact that the former is more stable compared with the latter at low temperatures. The CrB-type PtZr compound possesses two equivalent atom positions (coordination number=9 and 17 for atoms Pt and Zr, respectively) in one cell. The CrB-type phase is most stable, possessing the lowest formation enthalpy, in all the considered PtZr compounds. According to the reports on the Pt3Zr compound [26], two stable phases (Pm3m-Cu3Au and P63/mmc-Ni3Ti) are established in our ab initio calculation, and they are determined to possess quite close formation enthalpies, i.e. -106.15 and -106.97 kJ/mol, respectively. As shown in Table 3, the calculated lattice constants of the two stable phases are a=4.057 and a=5.745 , c=9.401 , consisting with those of the synthesized phases in experiment (a=3.99 and a=5.264 , c=9.213 ) [27]. The found of the Mn5Si3-type Pt3Zr5 phase with three equivalent atom positions in one cell was first reported by BISWAS and SCHUBERT [32]. The coordination numbers of the hexagonal structure are 14, 15 and 11 for atoms Zr1, Zr2 and Pt1, respectively. In our calculation, the lattice constants of Pt3Zr5 (Mn5Si3) are a=8.287 and c=5.416 , with a formation enthalpy of -86.72 kJ/mol. For the Pt11Zr9 compound, PANDA and BAHN [33] reported that they prepared a sample by annealing a cast sample at 1473 K for 1 h, which produced only diffraction lines of the tetragonal Pt11Zr9 structure (space group I4/m). In our calculation, the formation enthalpy of Pt11Zr9 compound with parameters of a=10.486 and c=6.856 , is much higher than that of other stable phases, indicating that some phase separation would occur at the composition of atom Pt-55% at low temperatures. Although there are rare reports on the Pt10Zr7 compound, it was selected as a stable phase in this study based on the research contribution by DWIGHT et al [27]. The Pt10Zr7 compound possesses an orthorhombic crystal structure (Ni7Zr10-type), with calculated (experimental) lattice parameters of a=9.739 (9.613) , b=9.701 (9.612) and c=13.330 (13.087) , presenting a consistency. According to the calculated results, Pt10Zr7 (Ni7Zr10) is proved to be a stable phase at low temperatures, since the formation enthalpy is quite negative. For the Pt8Zr (Pt8Ti) compound, it was first found by KRAUTWASSER et al [31], but there have been seldom reports about it.
, b=10.315 , c=4.277 ). The orthorhombic structure exits stably up to 1848 K, but then is replaced by a cubic B2-type phase with a=3.3851 at 1863��10 K [27, 34]. In experiment, the orthorhombic phase re-appeared on cooling, indicating that there is a typically martensitic transformation existing in the compound. As presented in Table 3, the calculated results show that the lattice parameters of the orthorhombic CrB-type phase are a=3.445 , b=10.436 , c=4.328 , and that of the B2-type phase is a=3.356 . The deviation of lattice constants is in the order of 0.01 or in the range of 1.17%, indicating that the calculated results are in accordance with the experimental ones. Simultaneously, it is found that the formation enthalpy of the orthorhombic phase is about 20 kJ/mol lower than that of B2-type phase at 0 K, providing a thermodynamic basis for the fact that the former is more stable compared with the latter at low temperatures. The CrB-type PtZr compound possesses two equivalent atom positions (coordination number=9 and 17 for atoms Pt and Zr, respectively) in one cell. The CrB-type phase is most stable, possessing the lowest formation enthalpy, in all the considered PtZr compounds. According to the reports on the Pt3Zr compound [26], two stable phases (Pm3m-Cu3Au and P63/mmc-Ni3Ti) are established in our ab initio calculation, and they are determined to possess quite close formation enthalpies, i.e. -106.15 and -106.97 kJ/mol, respectively. As shown in Table 3, the calculated lattice constants of the two stable phases are a=4.057 and a=5.745 , c=9.401 , consisting with those of the synthesized phases in experiment (a=3.99 and a=5.264 , c=9.213 ) [27]. The found of the Mn5Si3-type Pt3Zr5 phase with three equivalent atom positions in one cell was first reported by BISWAS and SCHUBERT [32]. The coordination numbers of the hexagonal structure are 14, 15 and 11 for atoms Zr1, Zr2 and Pt1, respectively. In our calculation, the lattice constants of Pt3Zr5 (Mn5Si3) are a=8.287 and c=5.416 , with a formation enthalpy of -86.72 kJ/mol. For the Pt11Zr9 compound, PANDA and BAHN [33] reported that they prepared a sample by annealing a cast sample at 1473 K for 1 h, which produced only diffraction lines of the tetragonal Pt11Zr9 structure (space group I4/m). In our calculation, the formation enthalpy of Pt11Zr9 compound with parameters of a=10.486 and c=6.856 , is much higher than that of other stable phases, indicating that some phase separation would occur at the composition of atom Pt-55% at low temperatures. Although there are rare reports on the Pt10Zr7 compound, it was selected as a stable phase in this study based on the research contribution by DWIGHT et al [27]. The Pt10Zr7 compound possesses an orthorhombic crystal structure (Ni7Zr10-type), with calculated (experimental) lattice parameters of a=9.739 (9.613) , b=9.701 (9.612) and c=13.330 (13.087) , presenting a consistency. According to the calculated results, Pt10Zr7 (Ni7Zr10) is proved to be a stable phase at low temperatures, since the formation enthalpy is quite negative. For the Pt8Zr (Pt8Ti) compound, it was first found by KRAUTWASSER et al [31], but there have been seldom reports about it.
Considering the relative thermodynamic stability of calculated phases, it is important to clarify whether high-energy phases are mechanically or dynamically stable in the bulk form. This issue can generally be assessed by analyzing single-crystal elastic constants. Basically in this study, by calculating the formation enthalpies as a function of appropriate lattice deformations, ab initio electronic-structure methods were used to compute single-crystal elastic constants, with the calculated results shown in Table 4. To obey actual situation to the great extent, the considered phases mainly include those Pt-Zr stable and hypothetical phases around the ground-state convex hull.
The internal energy (E(V, {��})) of a crystal under an infinitesimal strain ��, can be calculated as
 (1)
(1)
where V0 is the volume of the original crystal with corresponding energy E(V0,0), Cij are the single-crystal elastic constants; ��={ei, ej, ��} presents the members of strain tensor.
The elastic constants of stable phases should obey the mechanical stability conditions based on the Born Standard [35]. For cubic lattice symmetry, there are three single-crystal elastic constants, C11, C12, and C44, and the mechanical stability conditions are C11>0, C12>0, C44>0, C11-C12>0. Basically, stable PtZr (Pm3m) and hypothetical PtZr3 (Fm3m) phases with cubic structure both satisfy the criteria of mechanically stability, as shown in Table 4. Similarly, the requirements of mechanical stability in a tetragonal crystal are: C11>0, C12>0, C13>0, C33>0, C44>0, C66>0, C11-C12>0, 2C11+C33+2C12-4C13>0 and C11+C33-2C13>0 [36]. For the stable phases Pt11Zr9 (I4/m), Pt2Zr (I4/mmm) and Pt8Zr (I4/mmm), with tetragonal lattice symmetry, single- crystal elastic constants obey the above stability conditions. For hypothetical tetragonal phases, they satisfy the above conditions, indicating that any of these phases will be mechanically stable at intermediate compositions.
The requirements of mechanical stability in a hexagonal crystal are: C11-C12>0, C11+C12+C33>0, (C11+C12)C33> >0 and C44>0 [36]. The calculated phases, possessing hexagonal-structured lattice are all determined to be mechanically stable. For orthorhombic crystals, they all obey the mechanical stability conditions, except for two phases (Pt5Zr8 and Pt3Zr8 possess a negative C55 or C66), not satisfying the conditions: Cii>0 (i=1-6), C11+C22-2C12>0, C11+C22+C33+2C12+2C13+ 2C23>0 and C22+C33-2C23>0 [36]. Therefore, it is not mechanically stable for the hypothetical Pt5Zr8 (Pbam) and Pt3Zr8 (Pnma) phase, implying that it is unlikely that any of these phases will be mechanically unstable at intermediate compositions. According to the above analysis, the calculated phases mostly satisfy the criteria of mechanically stability, implying that it is unlikely that any of these phases will be mechanically unstable at intermediate compositions.
>0 and C44>0 [36]. The calculated phases, possessing hexagonal-structured lattice are all determined to be mechanically stable. For orthorhombic crystals, they all obey the mechanical stability conditions, except for two phases (Pt5Zr8 and Pt3Zr8 possess a negative C55 or C66), not satisfying the conditions: Cii>0 (i=1-6), C11+C22-2C12>0, C11+C22+C33+2C12+2C13+ 2C23>0 and C22+C33-2C23>0 [36]. Therefore, it is not mechanically stable for the hypothetical Pt5Zr8 (Pbam) and Pt3Zr8 (Pnma) phase, implying that it is unlikely that any of these phases will be mechanically unstable at intermediate compositions. According to the above analysis, the calculated phases mostly satisfy the criteria of mechanically stability, implying that it is unlikely that any of these phases will be mechanically unstable at intermediate compositions.
Accordingly, the calculated formation enthalpies are plotted as a function of Pt content, as shown in Fig. 1(a). The ground-state convex hull, consisting of Zr (P63/mmc), Pt3Zr5 (Mn5Si3), PtZr (CrB), Pt10Zr7 (Ni7Zr10), Pt3Zr (Ni3Ti), Pt8Zr (Pt8Ti) and Pt  is asymmetric and skewed a little towards Pt side. These phases are energetically favored to be formed, but the other factors, such as structures and mechanical properties are not included. As reported, three stable compounds, Pt3Zr, PtZr and Pt3Zr5, possess different melting points of 2427, 2377 and 2000 K, respectively. The information presented in reference provides an certification for the fact that our calculated formation enthalpies of Pt3Zr and PtZr are quite close, and that of Pt3Zr5 is significantly higher than the other two. As thermodynamically predicted, an eutectic reaction would occur at the composition around Pt11Zr9, because of its high formation enthalpy far from the ground-state hull, i.e., a phase separation Pt11Zr9
is asymmetric and skewed a little towards Pt side. These phases are energetically favored to be formed, but the other factors, such as structures and mechanical properties are not included. As reported, three stable compounds, Pt3Zr, PtZr and Pt3Zr5, possess different melting points of 2427, 2377 and 2000 K, respectively. The information presented in reference provides an certification for the fact that our calculated formation enthalpies of Pt3Zr and PtZr are quite close, and that of Pt3Zr5 is significantly higher than the other two. As thermodynamically predicted, an eutectic reaction would occur at the composition around Pt11Zr9, because of its high formation enthalpy far from the ground-state hull, i.e., a phase separation Pt11Zr9 Pt3Zr5+PtZr is expected to be observed in experiment. For Pt10Zr7 compound, it possesses a little higher formation enthalpy (-113.74 kJ/mol) than that of the most stable phase PtZr (CrB) (-115.10 kJ/mol), indicating that it is advantaged for Pt10Zr7 (Ni7Zr10) phase to be formed in thermodynamics. Actually, although there are rare reports on Pt10Zr7 (Ni7Zr10) compound, it has indeed been obtained early in 2007 [30]. Concerning the hypothetical compounds, it can be seen that the formation enthalpies of several phases are quite close to the ground-state convex hull as shown in Fig. 1(a). These compounds could thermodynamically be synthesized in practical experiment, and in contrary, those phases with extremely high formation enthalpies are hardly to be synthesized. It should be mentioned that some other researchers have also reported the ab initial calculated results of the Pt-Zr system. For example, XING et al [17] reported several calculated structures of PtZr compounds, and CURTAROLO et al [37] also presented a ground-state convex hull for the Pt-Zr system. The general trend of our results is similar with their data, despite there exist some differences in detail.
Pt3Zr5+PtZr is expected to be observed in experiment. For Pt10Zr7 compound, it possesses a little higher formation enthalpy (-113.74 kJ/mol) than that of the most stable phase PtZr (CrB) (-115.10 kJ/mol), indicating that it is advantaged for Pt10Zr7 (Ni7Zr10) phase to be formed in thermodynamics. Actually, although there are rare reports on Pt10Zr7 (Ni7Zr10) compound, it has indeed been obtained early in 2007 [30]. Concerning the hypothetical compounds, it can be seen that the formation enthalpies of several phases are quite close to the ground-state convex hull as shown in Fig. 1(a). These compounds could thermodynamically be synthesized in practical experiment, and in contrary, those phases with extremely high formation enthalpies are hardly to be synthesized. It should be mentioned that some other researchers have also reported the ab initial calculated results of the Pt-Zr system. For example, XING et al [17] reported several calculated structures of PtZr compounds, and CURTAROLO et al [37] also presented a ground-state convex hull for the Pt-Zr system. The general trend of our results is similar with their data, despite there exist some differences in detail.
In Fig. 1(b), the mean-volumes of phases close to the ground-state hull are presented as a function of Pt contents. The polygonal region shown in Fig. 1(b) is surrounded by Vegard Law line and an envelope connected by several stable phases (Pt-fcc, Pt8Zr-Pt8Ti, Pt3Zr5-Mn5Si3, Pt3Zr-Ni3Ti, Pt3Zr-Cu3Au and Zr-hcp). As shown in Fig. 1(b), most data locate in the region, and a greater deviation between the calculated results and Vegard Law line appears on the Pt side. Since the Pt-Zr system possesses high negative formation enthalpies, volume contractions can be aroused by alloying, forming the negative bias of volumes from Vegard Law, and the results are in coincidence with the data provided by STALICK and WATERSTRAT [30].
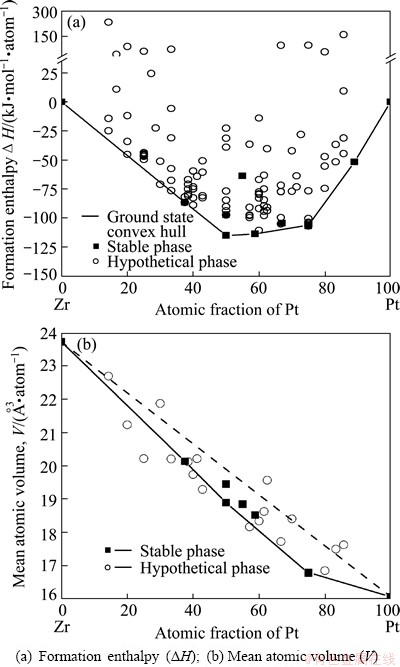
Fig. 1 Calculated structural properties of Pt-Zr compounds at zero temperature and pressure
4 Correlations of formation enthalpy/bulk modulus and volume
Based on the calculated results, the formation enthalpies are plotted as a function of differences between the calculated and ideal geometric atomic mean-volumes, as presented in Fig. 2(a). According to the calculated results of cell volume and atom numbers in unit cell, mean atomic volume can be obtained. In some extent, mean atomic volume can reflect average interatomic distance. To study the reflection of nonlinear relationship between interatomic force and distance, we focus on the two calculated property correlations: 1) formation enthalpy (��H) and volume difference (��V); 2) bulk modulus (B0) and mean atomic volume (V) for Pt-Zr compounds. As shown Fig. 2(a), the fitting line of the Pt-Zr system possesses a high slope and a low intercept, indicating that the increase of mean atomic volume can cause large elevation of formation enthalpy in some extent. From Fig. 2(a), it is also found that most points concentrated around (0, -50), indicating that negative formation enthalpy and contracted volume are advantaged factors for phase stability of Pt-Zr compounds. These two factors are not independent, i.e. volume reduction can induce formation enthalpy decline, and vice versa. Generally, over large volume increase or over high formation enthalpies would make it hardly for phases to be synthesized in structure and thermodynamics. Especially for stable compounds, the volume differences with respect to the ideal geometric atomic mean-volumes locate in a narrow region of -0.45 to -1.20 3 (corresponding to 2.8%-7.5% and 1.9%-5.1% with respect to Pt and Zr atomic mean-volume, respectively). Formation enthalpy and atomic volume are not independent, they are related to each other. Negative formation enthalpies and volume contraction can be favored for the formation of Pt-Zr compounds, and conversely, positive formation enthalpies and volume expansion would reduce the lattice stability of Pt-Zr compounds. Furthermore, volume contraction would bring the release of heat, which could be part of formation enthalpies, and simultaneously, larger formation enthalpies would draw atoms closer to each other, making atomic volume contracted. The reason of volume compressions can be resorted to the negative formation enthalpies and radius difference between Pt and Zr (15.1%). On one hand, different atoms tend to attract each other because of the interaction, directly making effective distances of atoms closer. On the other hand, atoms would become closer packed than that of ideally mechanical mixing due to size difference, leading to volume compressions of Pt-Zr compounds.

Fig. 2 Calculated property correlations of formation enthalpy (��H)-volume difference (��V) (a) and bulk modulus (B0)-mean atomic volume (V) (b) for Pt-Zr compounds
Considering the relationship of internal structure and external property, the calculated bulk modulus are plotted as a function of mean atomic volume, as shown in Fig. 2(b). Based on mechanical stability, only compounds with low formation enthalpies are included in this section. As shown in Fig. 2(b), black blocks represent the data of stable Pt-Zr compounds, and hollow circle for hypothetical ones. The points of metals Pt and Zr locate at two ends of the data region for reference. By fitting these data, there is a linear relationship with a negative slop (-0.18272) between the two factors, and these calculated points distribute quite evenly on both sides of the fitting line. Simultaneously, it can be seen that R2 is 0.55518, presenting that the factors for the Pt-Zr system have good relationship. It��s briefly concluded that bulk modulus increases linearly with reducing mean atomic volume. The larger the volume contraction becomes, the closer the distance of two atoms would be, making the atomic repulsion stronger. From a physical point of view, bulk modulus reflects the isotropic resistance of the volume compressions, which is the macro performance of atomic repulsion. As reported by GRIMVALL [38] that the volume dependence of elastic properties is closely related to their pressure dependence for the metals. The ratio of elastic constants and pressure can be expressed as a constant related to the sound velocity and mass densities of the metals. Since the Pt-Zr compounds with different compositions possess similar properties, it is supposed that their ratio of bulk modulus and mean atomic volume are quite close, resulting in an approximately linearly correlation of bulk moduli and mean atomic volumes, as shown in Fig. 2(b).
5 Conclusions
1) The lattice constants, cohesive energies and elastic properties of 118 compounds in the Pt-Zr system were obtained by ab initio calculation. The ground-state convex hull was also derived from the calculated formation energies, providing a basis for further thermodynamic calculations and atomistic simulations.
2) For these Pt-Zr compounds, a positive linear correlation between the formation enthalpies and atomic volumes, and a negative linear correlation between the bulk modules and atomic volumes were found.
References
[1] JIANG X W, LI S S, XIA J B, WANG L W. Quantum mechanical simulation of electronic transport in nanostructured devices by efficient self-consistent pseudopotential calculation [J]. J Appl Phys, 2011, 109: 054503.
[2] NAKANO H, YAMAMOTO T. Variational calculation of quantum mechanical/molecular mechanical free energy with electronic polarization of solvent [J]. J Chem Phys, 2012, 136: 134107.
[3] JIANG Y, XU C H, LAN G Q. First-principles thermodynamics of metal-oxide surfaces and interfaces: A case study review [J]. Transactions of Nonferrous Metals Society of China, 2013, 23: 180-192.
[4] NONG Z S, ZHU J C, YU H L, LAI Z H. First principles calculation of intermetallic compounds in FeTiCoNiVCrMnCuAl system high entropy alloy [J]. Transactions of Nonferrous Metals Society of China, 2012, 22: 1437-1444.
[5] WEN Y F, SUN J, HUANG J. First-principles study of stacking fault energies in Ni3Al intermetallic alloys [J]. Transactions of Nonferrous Metals Society of China, 2012, 22: 661-664.
[6] JUND P, VIENNOIS R, COLINET C, HUG G, FEVRE M, TEDENAC J C. Lattice stability and formation energies of intrinsic defects in Mg2Si and Mg2Ge via first principles simulations [J]. J Phys-Condens Mat, 2013, 25: 035403.
[7] ANDRIYEYSKY B, PILZ T, YEON J, HALASYAMANI P S, DOLL K, JANSEN M. DFT-based ab initio study of dielectric and optical properties of bulk Li2B3O4F3 and Li2B6O9F2 [J]. J Phys Chem Solids, 2013, 74: 616-623.
[8] HU H, REVEN L, REY A D. Ab initio DFT study of 6-mercapto-hexane SAMs: effect of Au surface defects on the monolayer assembly [J]. Mol Simulat, 2013, 39: 292-298.
[9] YANG L M, RAVINDRAN P, VAJEESTON P, TILSET M. Ab initio investigations on the crystal structure, formation enthalpy, electronic structure, chemical bonding, and optical properties of experimentally synthesized isoreticular metal-organic framework-10 and its analogues: M-IRMOF-10 (M= Zn, Cd, Be, Mg, Ca, Sr and Ba) [J]. RSC Adv, 2012, 2: 1618-1631.
[10] WANG W G, LI J H, DAI Y, LIU B X. Ab initio calculations to determine the formation enthalpy of Cu3Au phases [J]. Phil Mag Lett, 2010, 90: 801-807.
[11] LI C L, WANG Z Q, WANG C Y. Phase stability, mechanical properties and electronic structure of hexagonal and trigonal Ti5Al2C3: An ab initio study [J]. Intermetallics, 2013, 33: 105-112.
[12] ZHAO S Z, LI J H, LIU B X. Chemical and topological short-range orders in the ternary Ni-Zr-Al metallic glasses studied by Monte Carlo simulations [J]. J Phys-Condens Mat, 2013, 25: 095005.
[13] SHIN J H, KIM D I, CHO K M, SUEMATSU H, KIM K H, NOWAK R. Plasticity-induced nanocrystallization in a Zr65Cu35 thin film metallic glass [J]. J Non-Cryst Solids, 2013, 362: 65-68.
[14] AN Q, SAMWER K, GODDARD W A, JOHNSON W L, JARAMILLO-BOTERO A, GARRET G, DEMETRIOU M D. Predicted optimum composition for the glass-forming ability of bulk amorphous alloys: Application to Cu-Zr-Al [J]. Journal of Physical Chemistry Letters, 2012, 3: 3143-3148.
[15] IQBAL M, QAYYUM A, AKHTER J I. Surface modification of Zr-based bulk amorphous alloys by using ion irradiation [J]. J Alloys Compd, 2011, 509: 2780-2783.
[16] TAGUE M E, GREGOIRE J M, LEGARD A, SMITH E, DALE D, HENNIG R, DISALVO F J, VAN DOVER R B, ABRUNA H D. High throughput thin film Pt-M alloys for fuel electrooxidation: Low concentrations of M (M = Sn, Ta, W, Mo, Ru, Fe, In, Pd, Hf, Zn, Zr, Nb, Sc, Ni, Ti, V, Cr, Rh) [J]. J Electrochem Soc, 2012, 159: F880-F887.
[17] XING W W, CHEN X Q, LI D Z, LI Y Y, FU C L, MESCHEL S V, DING X Y. First-principles studies of structural stabilities and enthalpies of formation of refractory intermetallics: TM and TM3 (T= Ti, Zr, Hf; M=Ru, Rh, Pd, Os, Ir, Pt) [J]. Intermetallics, 2012, 28: 16-24.
[18] http://www.castep.org/.
[19] PERDEW J P, RUZSINSZKY A, CSONKA G I, VYDROV O A, SCUSERIA G E, CONSTANTIN L A, ZHOU X L, BURKE K. Restoring the density-gradient expansion for exchange in solids and surfaces [J]. Phys Rev Lett, 2008, 100: 136406.
[20] WHITE J A, BIRD D M. Implementation of gradient-corrected exchange-correlation potentials in car-parrinello total-energy calculations [J]. Phys Rev B, 1994, 50: 4954-4957.
[21] PFROMMER B G, COTE M, LOUIE S G, COHEN M L. Relaxation of crystals with the quasi-Newton method [J]. J Comput Phys, 1997, 131: 233-240.
[22] EYERT V. A comparative study on methods for convergence acceleration of iterative vector sequences [J]. J Comput Phys, 1996, 124: 271-285.
[23] LIDE D R. CRC handbook of chemistry and physics [M]. New York: CRC Press, 2002.
[24] DINSDALE A T. SGTE data for pure elements [J]. CALPHAD, 1991, 15: 317-425.
[25] HOLMES N C, MORIARTY J A, GATHERS G R, NELLIS W J. The equation of state of platinum to 660GPa (6.6mbar) [J]. J Appl Phys, 1989, 66: 2962-2967.
[26] RAMAN A, SCHUBERT K. Structural investigations on some alloy systems homologous and quasihomologous to T4-T9 [J]. Z Metallkd, 1964, 55: 704-710.
[27] DWIGHT A E, CONNER R A, DOWNEY J W. Equiatomic compounds of transition and lanthanide elements with Rh Ir Ni and Pt [J]. ACTA Cryst, 1965, 18: 835-839.
[28] BREWER L, WENGERT P R. Transition-metal alloys of extraordinary stability-example of generalized lewis-acid-base interactions in metallic systems [J]. Metall Trans A, 1973, 4: 83-104.
[29] GACHON J C, CHARLES J, HERTZ J. Different ways to find the thermodynamic functions describing the formation of binary-alloys .1. Comparison between models and experimental-data [J]. Calphad, 1985, 9: 29-34.
[30] STALICK J K, WATERSTRAT R M. The zirconium-platinum phase diagram [J]. J Alloys Compd, 2007, 430: 123-131.
[31] KRAUTWAS P, BHAN S, SCHUBERT K. Structural investigations in systems Ti-Pd and Ti-Pt [J]. Z Metallkd, 1968, 59: 724-729.
[32] BISWAS T K, SCHUBERT K. Some new phases of Mn5Si3 type [J]. Z Metallkd, 1967, 58: 558-559.
[33] PANDA S C, BHAN S. Crystal-structure of Zr9Pt11 [J]. J Less-Common Met, 1974, 34: 344-347.
[34] WODNIECKI P, WODNIECKA B, KULINSKA A, UHRMACHER M, LIEB K P. Temperature behavior of ZrPt compound studied by PAC with Ta-181 and Cd-111 probes [J]. J Alloys Compd, 2005, 399: 52-56.
[35] SIMMONS G, WANG H. Single crystal elastic constants and calculated aggregate properties: A handbook [M]. Cambridge: MIT Press, 1971.
[36] WALLACE DC. Thermodynamics of crystals [M]. New York: Wiley, 1972.
[37] CURTAROLO S, MORGAN D, CEDER G. Accuracy of ab initio methods in predicting the crystal structures of metals: A review of 80 binary alloys [J]. CALPHAD, 2005, 29: 163-211.
[38] GRIMVALL G. Thermophysical properties of materials [M]. Amsterdam: North-Holland, 1999.
�� ѩ1����Һ�1���� Ҷ2��������1
1. �廪��ѧ ����ѧԺ���Ƚ�����ʵ���ң����� 100084��
2. �й���ѧԺ �Ϻ�Ӧ�������о������Ϻ� 200240
ժ Ҫ��ͨ����һ��ԭ���ļ��㷽�����о�118�ֲ�ͬ�ṹ��Pt-Zr�м仯�����ѡȡ��ص��������ܣ���ṹ�ȶ��ԡ��������������ʡ����Գ����Լ���ģ���Ƚ��м��㡣���ݼ���ó�����������Ϣ������Pt-Zrϵͳ�Ļ�̬�������ߡ�����õ������������ϢΪδ��������ѧ�����ԭ�ӳ߶�ģ���ṩ�������ݡ���ѡȡ��Pt-Zr�������У���������������ع�ϵ����������ԭ���������������أ�����ģ����ԭ������ɸ�������ع�ϵ��
�ؼ��ʣ�Pt-Zrϵͳ����ѧ���ܣ��������ܣ���һ��ԭ�����㣻�����ʣ���ģ����ԭ�����
(Edited by Chao WANG)
Foundation item: Projects (50971072, 51131003) support by the National Natural Science Foundation of China; Projects (2011CB606301, 2012CB825700) supported by the Ministry of Science and Technology of China; Project supported by the Administration of Tsinghua University
Corresponding author: Bai-xin LIU; Tel: +86-10-62772557; Fax: +86-10-62771160; E-mail: dmslbx@tsinghua.edu.cn
DOI: 10.1016/S1003-6326(13)62920-9

