文章编号:1004-0609(2010)04-0743-06
Au-Cu系金属间化合物价电子结构及晶体结合能计算
蒋淑英,李世春
(中国石油大学(华东) 机电工程学院,东营257061)
摘 要:运用固体与分子经验电子理论,计算Au-Cu系金属间化合物的价电子结构、共价键键能及晶体理论结合能。计算结果表明:金属间化合物Au3Cu、AuCu、AuCu3的最强键分别为Au―Au键、AuⅠ―AuⅡ键、Au―Cu键,最强键键能分别为10.728 6、10.038和10.163 0 kJ/mol,晶体理论结合能分别为401.25、363.64和381.02 kJ/mol。用EET理论计算的晶体结合能值与用特征晶体理论计算的晶体结合能值基本吻合。3种化合物中,Au3Cu的最强键键能和晶体理论结合能最大,因此其稳定性最好。
关键词:Au-Cu体系;金属间化合物;经验电子理论;价电子结构;结合能
中图分类号:O641.1 文献标志码:A
Calculation of valence electron structures and cohesive energies of intermetallic compounds in Au-Cu system
JIANG Shu-ying, LI Shi-chun
(School of Electromechanical Engineering, China University of Petroleum (Huadong), Dongying 257061, China)
Abstract: The valence electron structures and cohesive energies of Au-Cu system intermetallic compounds were calculated based on the empirical electron theory of solids and molecules (EET). The results show that the strongest bonds of Au3Cu, AuCu and AuCu3 are Au―Au bond, AuⅠ―AuⅡ bond and Au―Cu bond, respectively, the energies of the strongest bonds are 10.728 6, 10.038 and 10.163 0 kJ/mol, respectively, and the theoretical cohesive energies are 401.25, 363.64 and 381.02 kJ/mol, respectively. The cohesive energy values calculated with EET theory are roughly consistent with those calculated by characteristic crystal theory. Among the three compounds, Au3Cu has the strongest bond energy and the biggest cohesive energy, therefore its stability is the best.
Key words: Au-Cu system; intermetallic compound; empirical electron theory; valence electron structure; cohesive energy
1978年,余瑞璜在能带理论、价键理论、电子浓度理论的基础上,提出了“固体与分子经验电子理论”(EET)[1],根据“键距差(BLD)法”,可以计算复杂化合物晶体的电子结构。对于晶体结构已知的晶体,EET根据原子杂化态能给出晶体键络的电子分布,再根据这些价电子结构信息,可以估算晶体的各种性能。这已被广泛应用于材料计算和材料设计[2-7],在理论和应用方面,积累了有用的数据。
Au-Cu体系是重要的基础合金系,其有序无序转变已经有较多的研究报道[8-14],而有关化合物价键电子结构研究的报道不多。YU等[15]根据以热力学为基础的特征晶体理论研究了Au-Cu系无序合金及有序金属间化合物的电子结构及晶体结合能。本文作者应用EET理论计算Au-Cu系主要金属间化合物的价电子结构、共价键键能和晶体理论结合能,并与文献[15]中应用特征晶体理论的计算结果进行比较。
1 计算方法
固体与分子经验电子理论的核心是原子的杂化状态和键距差法(BLD),键距差法利用已知的晶格参数求得晶体中各原子的杂化状态和它们之间的共价电子分布,建立晶体或分子的价电子结构。进行BLD计算的前提是必须知道其晶体结构,即必须知道晶体结构类型、晶格常数和原子坐标参数的具体数值。BLD计算中所用的基本理论工具是EET给出的共价键的键距公式:
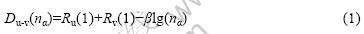
式中:脚标u和v表示成键的两个原子;Du-v(nα)表示u、v两个原子间的共价键距;nα表示u、v两个原子间的共价电子对数;α表示键序的标号,一般按键的长短从最短键开始依次标记为1,2,3,…;Ru(1)和Rv(1)分别表示u、v原子的单键半径;β是一个参数,其取值依赖于所讨论的分子或晶体中最强键的值[16]。
在同一体系内,所有由共价键连接的原子之间的键距都遵守键距公式。假设在结构单元中有N个不同长度的键,其键距分别为D(nα),对于每一共价键都有一个键距方程式。设最短键的α=1,其键距方程为
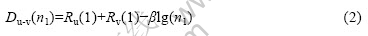
设其他任一键的标号为α′,且α′=2,3,4,…,N,则其键距方程为
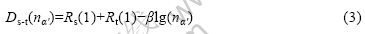
将D(n1)分别与D(n2),D(n3),…,D(nN)每个方程式的两边相减,则可得到N-1个键距差方程式:
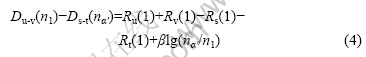
式中:脚标u和v、s和t表示两两成键的原子,u、v、s、t可以相同,也可以不同。
令

此外,考虑分子或晶体的基本结构单元呈电中性,因此,在一个结构单位中的各个原子所贡献出来的全部共价电子应该完全分配在该结构单元内的全部共价键上,从而可建立一个n1方程式:

式中:Iα表示一个结构单元中α键的等同键数; nc表示晶体中原子的共价电子数。
式(7)与上述N-1个γα′方程一起构成一个包含N个未知数和N个方程式的方程组。根据已知的原子杂化表,可以对所涉及原子的各个杂阶的单键半径R(1)、共价电子数nc代入方程组进行试算,这样对不同原子的每个杂阶组合可以计算出相应的nα值。 将nα值代入键距方程,得出理论键距D(nα)。当所取的杂阶符合原子所处的实际状态时,计算出的理论键距D(nα)应与实验键距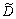 (nα)一致,根据经验,在一级近似下,两者之差的绝对值小于0.005 nm即满足条件。
(nα)一致,根据经验,在一级近似下,两者之差的绝对值小于0.005 nm即满足条件。
2 晶体结构及键络分布
2.1 晶体结构
根据文献[17]可知,室温下能够稳定存在的Au-Cu系金属间化合物有Au3Cu、AuCu、AuCu3,本研究应用EET理论对这3种金属间化合物的价电子结构进行了计算分析。
Au3Cu晶体具有有序面心立方结构,空间群为Pm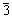 m(221),晶格常数为a=0.395 0 nm,每个晶胞中有3个Au原子和1个Cu原子,Au原子占据6个面心位置,Cu原子占据8个顶角位置,其晶体结构如图1所示。
m(221),晶格常数为a=0.395 0 nm,每个晶胞中有3个Au原子和1个Cu原子,Au原子占据6个面心位置,Cu原子占据8个顶角位置,其晶体结构如图1所示。
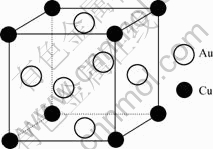
图1 Au3Cu的晶体结构
Fig.1 Crystal structure of Au3Cu
AuCu晶体具有有序面心立方结构,空间群为P4/mmm(123),晶格常数为a=0.397 5 nm,b=0.397 5 nm,c=0.368 5 nm,每个晶胞中有2个Au原子和2个Cu原子,Cu原子占据4个面心位置,Au原子占据8个顶角位置和2个面心位置,3种结晶学不等效原子AuⅠ、AuⅡ和Cu分别占据a、c和e乌科夫等效位置,其晶体结构如图2所示。
AuCu3晶体具有有序面心立方结构,空间群为Pmm(221),晶格常数为a=0.374 7 nm,每个晶胞中有1个Au原子和3个Cu原子,Au原子占据8个顶角位置,Cu原子占据6个面心位置,其晶体结构如图3所示。
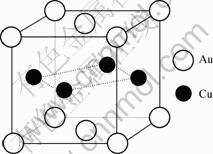
图2 AuCu的晶体结构
Fig.2 Crystal structure of AuCu
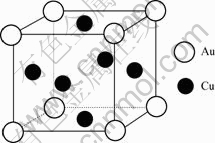
图3 AuCu3的晶体结构
Fig.3 Crystal structure of AuCu3
2.2 键络分布
通常原子的单键半径只有0.1 nm左右,当原子间距比2个原子单键半径大很多时,两者不会形成共价键。根据一般经验,2个原子的间距超过0.4 nm时,其成键作用已经可忽略,为谨慎起见,实际计算成键作用时,原子的间距可到0.5 nm左右[16]。基于此原则,Au3Cu中不可忽略的键有5种,AuCu中不可忽略的键有12种, AuCu3中不可忽略的键有5种。根据晶体结构、晶格常数和各原子坐标参数值计算得到Au3Cu、AuCu和AuCu3晶胞中各键的实验键距(nα)及等同键数Iα分别见表1、表2和表3所列。
3 价电子结构计算
根据张瑞林[16]给出的原子杂化状态表,Au原子有18种杂化状态,Cu原子有18种A型杂化态和18种B型杂化态。将Au的18种杂阶(σ)与Cu的18种A型杂阶进行组合试算,根据EET判据:|(nα)-D(nα)|<0.005 nm,符合条件的杂阶组合就是晶体中可能存在的原子状态。当键距差小于0.005 nm时,理论键距D(nα)与实验键距(nα)在一级近似范围内是一致的。
将价电子结构计算过程用C语言编程,并经计算机扫描计算,得到晶体中每条不可忽略的共价键的理论键距、共价电子数和键距误差。在Au3Cu的计算结果中,满足键距差判据的杂阶组合(σN)共有208组;在AuCu的计算结果中,满足键距差判据的杂阶组合共有239组;在AuCu3的计算结果中,满足键距差判据的杂阶组合共有224组。一般情况下,键距差最小的状态为化合物最稳定的状态,因此,选取键距差最小的状态确定化合物的价电子结构,其计算结果如表1~3所列。从计算结果看,3种化合物的键距误差都远小于0.005 nm,说明理论键距与实验键距十分吻合,所选原子状态为晶体中原子实际存在的状态。
4 键能及理论结合能计算
4.1 键能计算
对于同种原子共价键的键能计算,按照EET理
表1 Au3Cu键络分布及价电子结构
Table 1 Bond nets and valence electron structures of Au3Cu
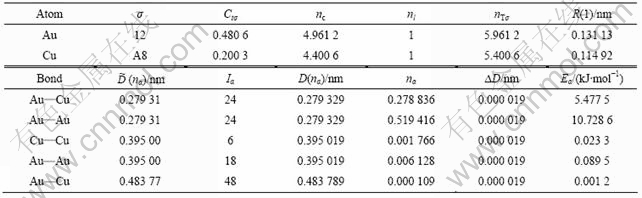
表2 AuCu键络分布及价电子结构
Table 2 Bond nets and valence electron structures of AuCu
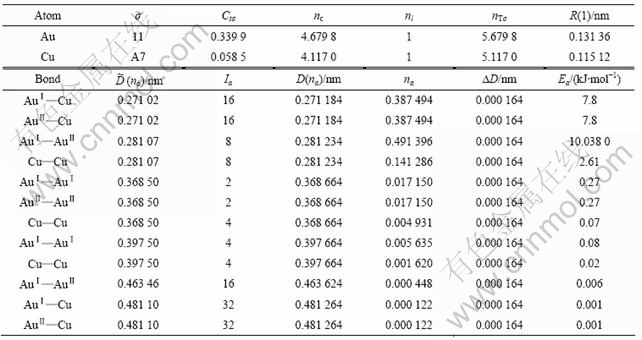
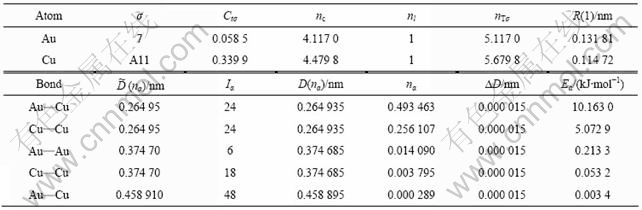
表3 AuCu3键络分布及价电子结构
Table 3 Bond nets and valence electron structures of AuCu3
论[16],其共价键键能公式为

式中:Eα为α键的共价键键能;nα为α键上的共价电子数;D(nα)为α键的理论键距;b为电子对核电荷的屏蔽作用系数,取值由文献[16]中的表6-4给出;f为α键上共价电子的成键能力,对于s-p-d型杂化,其计算式为

式中:a、β和γ值由原子的杂化态决定,其计算式分别为


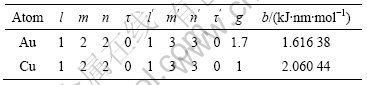
式中:l、m、n和l′、m′、n′分别表示h态和t态s、p、d共价电子和晶格电子数;τ和τ′是同一个参数;Ctσ表示原子σ杂阶中t态的成分;nTσ表示原子在σ杂阶时的总价电子数;g是反映d电子的自旋-轨道耦合效应对成键能力的贡献,对于第4、5、6周期的元素,g的取值分别为1.00、1.35、1.70。
根据文献[16]提供的原子状态杂化表,Au和Cu 原子h态与t态的s、p、d共价电子数、晶格电子数、τ参数及b值如表4所列。Au和Cu分别属于第6周期和第4周期元素,因此,g值分别为1.70和1.00。
表4 原子状态参数表
Table 4 State parameters of atoms
对于由两个不同原子形成的共价键键能计算,徐万东等[18]首先导出了其计算公式:

根据前面价电子结构计算所确定的原子状态以及共价键键能计算公式,3种化合物晶体中不可忽略的共价键键能Eα值如表1~3所列。根据表中计算结果:Au3Cu的最强键是Au―Au键,其键能为10.728 6 kJ/mol;AuCu的最强键是AuⅠ―AuⅡ键,其键能为10.038 0 kJ/mol;AuCu3的最强键是Au―Cu键,其键能为10.163 0 kJ/mol。因此,Au3Cu的最强键的键能最大,可以预见其稳定性最好。
4.2 理论结合能计算
根据文献[18-20],过渡金属化合物晶体结合能计算公式为
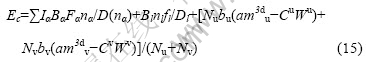
式(15)中的第一项反映了共价电子对结合能的贡献,第二项反映了晶格电子对结合能的贡献,第三项反映了磁电子和哑对电子对结合能的贡献。由于Au和Cu原子的价电子中不含磁电子,且哑对电子项中的C参数为零,因此磁电子和哑对电子对结合能的贡献为零。
对于Au-Cu系化合物晶体,其结合能计算公式为
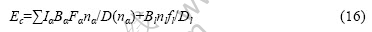
式中:Iα、Bα、Fα、nα、D(nα)、bu和bv所表示的意义与前面相同;Bl表示晶体结构式中晶格电子的屏蔽系数,Bl =(bumbvn)1/ (m+n),m和n为化合物分子式中包含的u原子和v原子的原子数;Dl为加权平均键长,Dl=ΣIα D(nα)/ΣIα;nl表示晶格电子数;fl表示晶格电子的成键能力,fl =(2nl/nT)1/2,nT为总价电子数。
将前面所确定的3种化合物的价电子结构参数带入结合能计算公式,得到Au3Cu、AuCu和AuCu3的理论结合能分别为401.25、363.64和381.02 kJ/mol。文献[15]中用特征晶体理论计算了Au3Cu、AuCu和AuCu3的结合能,其值分别为365.40、360.12和351.06 kJ/mol。对比两种方法的计算结果,可见前者的计算值比后者的计算值大,偏差分别为8.9%、1%、7.8%,均小于10%,两种计算方法得出的结果在一级近似下基本吻合。这说明本研究在计算3种化合物的价电结构中,所取原子杂阶正确,同时验证了EET理论和特征晶体理论的相容性。
5 结论
1) 在Au3Cu的价电子结构中,Au原子的杂阶为12,Cu原子的杂阶为A8;最强健为Au―Au键,其上共价电子数为0.519 4,键能为10.728 6 kJ/mol;晶体理论结合能为401.25 kJ/mol。
2) 在AuCu的价电子结构中,Au原子的杂阶为11,Cu原子的杂阶为A7;最强健为AuⅠ―AuⅡ键,其上共价电子数为0.491 4,键能为10.038 0 kJ/mol;晶体理论结合能为363.64 kJ/mol。
3) 在AuCu3的价电子结构中,Au原子的杂阶为7,Cu原子的杂阶为A11;最强健为Au―Cu键,其上共价电子数为0.493 5,键能为10.163 0 kJ/mol;晶体理论结合能为381.02 kJ/mol。
4) 3种化合物中,Au3Cu的最强键键能和晶体理论结合能最大,可以预见其稳定性最好。
REFERENCES
[1] 余瑞璜. 固体与分子经验电子理论[J]. 科学通报, 1978, 23(4): 217-224.
YU Rui-huang. Empirical electron theory in solids and molecules[J]. Chinese Science Bulletin, 1978, 23(4): 217-224.
[2] 刘志林, 孙振国, 李志林. 余氏理论和程氏理论在合金研究中的应用[J]. 自然科学进展, 1998, 8(2): 150-160.
LIU Zhi-lin, SUN Zhen-guo, LI Zhi-lin. Application of EET and TED in research of alloys[J]. Progress in Natural Science, 1998, 8(2): 150-160.
[3] 杜晓东, 丁厚福, 宣天鹏. CrB价电子结构对其性能的影响[J]. 中国有色金属学报, 2005, 15(12): 1980-1985.
DU Xiao-dong, DING Hou-fu, XUAN Tian-peng. Effect of valence electron structure on property of CrB[J].The Chinese Journal of Nonferrous Metals, 2005, 15(12): 1980-1985.
[4] 高英俊, 钟夏平, 刘 慧, 吴伟明. 微量Sc对Al-Mg合金晶粒细化影响的电子结构分析[J]. 中国有色金属学报, 2002, 12(S2): 132-134.
GAO Ying-jun, ZHONG Xia-ping, LIU Hui, WU Wei-ming. Valence electron structures of Al-Mg alloy with miner Sc and its effect on grain refinement[J]. The Chinese Journal of Nonferrous Metals 2002, 12(S2): 132-134.
[5] 李 文, 关振中, 杜立明, 支 文, 殷方信, 张瑞林, 余瑞璜. Ti-Al系金属间化合物价电子结构对其脆性的影响[J]. 金属热处理, 1996, 21(11): 3-5.
LI Wen, GUAN Zheng-zhong, DU Li-ming, ZHI Wen, YIN Fang-xin, ZHANG Rui-lin, YU Rui-huang. Research of valence electron structures and embrittlement of titanium-aluminides system[J]. Heat Treatment of Metals, 1996, 21(11): 3-5.
[6] 李 文, 张瑞林. 金属间化合物的价电子结构脆性判据[J]. 长春大学学报, 1999, 9(1): 11-15.
LI Wen, ZHANG Rui-lin. A valence electron structure criterion of embrittlement of intermetallics[J]. Journal of Changchun University, 1999, 9(1): 11-15.
[7] 张建民, 张瑞林, 余瑞磺, 郑伟涛, 胡安广. Fe-Al合金有序无序相变的电子理论研究[J]. 金属学报, 1994, 30(5): 204-210.
ZHANG Jian-min, ZHANG Rui-lin, YU Rui-huang, ZHANG Wei-tao, HU An-guang. Electron theory study of order-disorder transformations in Fe-Al alloys[J]. Acta Metallurgica Sinica, 1994, 30(5): 204-210.
[8] OKAMOTO H, CHAKRABARTI D J, LAUGHLIN D E, MASSAlSKI T B. The Au-Cu(gold-copper) system[J]. Bull Alloy Phase Diagram, 1987, 8(5): 454-473.
[9] CLAESON T, BOYCE J B. Order-disorder transformation in Au-Cu alloys studied by extended x-ray-absorption fine structure[J]. Phys Rev, 1984, B29: 1551-1557.
[10] SHOCKLEY W. Order-Disorder transformation in Au-Cu alloy[J]. J Chem. Phys, 1938, 6: 130-138.
[11] SYKES C, EVANS H. The transformation in the copper-gold alloy CU3Au[J]. J Inst Met, 1936, 58: 255-281.
[12] TOPOR L, KLEPPA O J. Thermochemistry of binary liquid gold alloys[J]. Metal Trans A, 1984, 15: 203-208.
[13] ORIANI R A. Thermodynamics of liquid Ag-Au and Au-Cu alloys[J]. Acta Metall, 1954, 2: 15-25.
[14] Orr R L. Heats of formation of solid Au-Cu alloys[J]. Acta Metall, 1960, 8(7): 489-493.
[15] YU Fang-xin, XIE You-qing, NIE Yao-zhuang. Electronic structure of Au-Cu alloys[J]. Trans Nonferrous Met Soc China, 2004, 14(6): 1041-1049.
[16] 张瑞林. 固体与分子经验电子理论[M]. 吉林: 吉林科学技术出版社, 1993.
ZHANG Rui-lin. Empirical electron theory of solid and molecules[M]. Jilin: Jilin Science and Technology Press, 1993.
[17] BRANDES E A, BROOK G B. Smithells metals reference book[M].7th ed. Burlingtom: Elsevier Butterworth-Heinemann Ltd, 1992.
[18] 徐万东, 张瑞林, 余瑞璜. 过渡金属化合物晶体结合能计算[J]. 中国科学A, 1988, 18(3): 323-330.
XU Wang-dong, ZHANG Rui-lin, YU Rui-huang. Calculation of cohesive energy of transition metal compounds[J]. Science in China A, 1988, 18(3): 323-330.
[19] 陈舜麟, 顾 强, 王天民. Co3Ti与CoTi的晶体结构与结合能的计算及其脆性[J]. 物理学报, 1995, 44(6): 936-942.
CHEN Shun-lin, GU Qiang, WANG Tian-ming. Calculation of cohesive energy and lattice constants of the intermetallic compounds Co3Tiand CoTi and their brittleness[J]. Acta Physica Sinica, 1995, 44(6): 936-942.
[20] 贾 堤, 董治中, 于申军, 刘文西. TiMe合金的价电子结构分析及结合能计算[J]. 稀有金属材料与工程, 1998, 27(3): 152-155.
JIA Di, DONG Zhi-zhong, YU Shen-jun, LIU Wen-xi. Calculation of cohesive energies and analysis of valence electronic structures of TiMe alloys[J]. Rare Metal Materials and Engineering, 1998, 27(3): 152-155.
(编辑 何学锋)
基金项目:国家自然科学基金资助项目(50371059)
收稿日期:2009-05-25;修订日期:2009-09-15
通信作者:蒋淑英,讲师,博士研究生;电话:0546-8393907;E-mail:jsy0430@gmail.com

