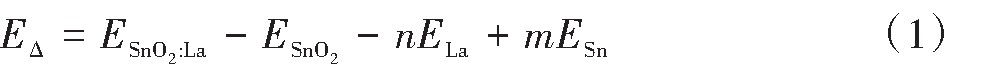������ʱ��: 2019-01-28 07:00
ϡ�н��� 2020,44(09),934-940 DOI:10.13373/j.cnki.cjrm.xy18110024
La����AgSnO2 ��ͷ���������ܵĵ�һ��ԭ���о�
���� ������ ���� ��˫�� ����
�ӱ���ҵ��ѧʡ�������繤װ���ɿ��������ܻ������ص�ʵ����
�ӱ���ҵ��ѧ�ӱ�ʡ��ų�������ɿ����ص�ʵ����
ժ Ҫ��
���ϡ�н���Laԭ�Ӳ��Ӷ�AgSnO2��ͷ���������ܵ�Ӱ�죬�����ܶȷ������ۡ���һ��ԭ���Լ�CASTEP�����Բ�����ϵ����ѧ���ʽ��������ۼ��㣬�õ��˲���ǰ��SnO2������γ��ܡ������ס�����̬�ܶȼ��ֲ�̬�ܶȺ������ѧ���������Ը�������������ͼ���������۷�����������������Ӳ�ͬŨ�ȵ�Laԭ�Ӻ���ϵ���γ��������нϴ��𣬲�������������ߣ�������Ũ��Ϊ25%ʱ��������δ������Ƶ��˵����Ũ���µIJ�����ϵ���ȶ����ڡ�ͨ������̬�ܶ�ͼ���ֲ�̬�ܶ�ͼ���Եõ�����ϵ��Laԭ����Oԭ�Ӿ��н�ǿ��������ã���Laԭ�ӵIJ��ӿ�����Ч�ؼ���Oԭ�ӵĻ��ԣ�ʹ�μ����˶��ķ����������࣬�����ݽ�δ������ϵ�õ�������ߡ�La�IJ���ʹSnO2������ء������ܼ������߱�����Զ��ͣ�����ϵ�Ļ��Ҷ����ӣ����������ܽ��ͣ����յ��������ӣ��Ӷ�����˾���Ļָ����������Բ����ȵ��ʵĴ�С�����ϴ��Ӱ�졣
�ؼ��ʣ�
��һ��ԭ�� ;La����SnO ;������ ;����ѧ���� ;
��ͼ����ţ� O469
����飺 ���1990-�����У��ӱ������ˣ�˶ʿ�о������о���������Ӵ����ϣ�E-mail:963601593@qq.com��; *�����ۣ����ڣ��绰��022-60204354,E-mail:jqwang@hebut.com;
�ո����ڣ� 2018-11-25
���� ������Ȼ��ѧ������Ŀ(51777057)����;
First-Principles Study on Thermal Properties of La-doped AgSnO2 Contact Materials
Chen Ling Wang Jingqin Liu Zhou Yu Shuangmiao Zhu Yancai
State Key Laboratory of Reliability and Intelligence of Electrical Equipment,Hebei University of Technology
Key Laboratory of Electromagnetic Field and Electrical Apparatus Reliability of Hebei Province,Hebei University of Technology
Abstract��
According to the influence of La atom doping on the thermal properties of AgSnO2 contact materials,theoretical calculations were carried out by density functional theory,first-principle and CASTEP software. The formation energy,phonon spectrum,phonon state density and partial wave state density and related thermodynamic parameters of the SnO2 crystal before and after doping were obtained,and the calculation results and curves were analyzed theoretically. The results showed that the doped system had good forming ability. Combined with the phonon spectrum curve,when the doping concentration was 25%,the phonon spectrum did not show a virtual frequency,and the crystal could exist stably. Through the analysis of density of states and the density of partial wave states,La atoms and O atoms had a strong coupling effect,which effectively stimulated the activity of O atoms,which increased the number of molecules participating in motion,that was,the heat capacity was significantly improved compared with the undoped system. The doping of La made the entropy,free energy and enthalpy curve of the SnO2 crystal became steep,that was,the chaos of the system increased,the free energy of the crystal decreased,and the absorbed heat increased,which improved the recovery ability of the crystal and it also had a large effect on the thermal conductivity of the material.
Keyword��
first-principle; La-doped SnO2 ; vibration property; thermodynamic property;
Received�� 2018-11-25
Ag Sn O2 ������һ�����͵ľ�����ʴ�ԡ���ĥ���ԡ�ʹ���������ĵ������ش�ͷ����
[1 ]
������Ag Sn O2 �����нӴ�����������ߡ��ӹ����ѵ�ȱ����������ʹ�á���Ҫԭ������Sn O2 ��һ�ֿ������뵼����ϣ�������Ե����ʹ�ò��ϵĵ������������ϸߣ��Ե��硢���������谭����
[2 ,3 ]
���о��������ھ������������ʡ�ȱ�ݻ������ѧ�����ȵ�ƫ�룬���п��ܸ��Ʋ��ϵĵ������ܺ���ѧ����
[4 ]
��
��ǰ���������о�����������Ag Sn O2 ��ͷ���ϵĵ��ӽṹ���ܴ��ṹ����ɲ��Ӽ���ѧ���ܵȷ��棬��ȱ�����侧����ѧ���о�����������
[5 ]
�о�����������������֮�������ã������Ź�����ϵĵ絼���������ȵ������ԡ������������������ص�������ѧ����ؽ������̽��������Sn O2 ���������ȶ��ԡ���ѧ���ʣ��羧�����ݣ����������ʣ����ȴ����������������¶�ЧӦ���������ͣ���
[6 ]
���������Ǿ�����ѧ�Ļ�������ӳ���Dz����ڲ��ĸ�����ģʽ�����������ֱ�ӷ�ӳԭ�Ӽ������ã��ǹ����������ѧ������Ħ�����ݡ��°��¶��Լ�������ϵ���Ļ������������ݣ����¼�����ݣ����о�����������Ҫ������ѧ���������������ܵ������������ͨ��������ֱ�Ӽ���õ�
[7 ]
��
�������û����ܶȷ������ۣ�density func�\tional theory,DFT���ĵ�һ��ԭ�������Բ�ͬŨ��La���ӵ�Sn O2 ��ͷ���ϵĸ������������м���
[8 ,9 ]
���õ��γ��ܣ��жϳ���ϵ�γɵ����׳̶ȡ�����������ף���þ������������������߽��������жϾ���ṹ���ȶ��ԡ�Ȼ�����ԭ��֮������÷���ϡ��Ԫ��La����ʹ��Sn O2 ����ѧ���ʵõ����ơ���������̬�ܶȻ�þ����������¶ȵı仯���ߣ����������ӶԾ����ȵ��ʵ�Ӱ�졣
ͨ����һ��ԭ���ļ��㣬�������ڶ���������ɸѡ�����ֽ��ŵIJ�����ϵ����СѰ�ҷ�Χ��Ϊ�ҵ��ϺõĴ�������ṩ��ָ��������ͨ���ҵ��ĺ��ʲ�����Ƚ��д�������Ʊ����������������鷽����о�����Ч�Ľ�ʡ��������������������
1����ģ�ͺͼ��㷽��
1.1����ģ��
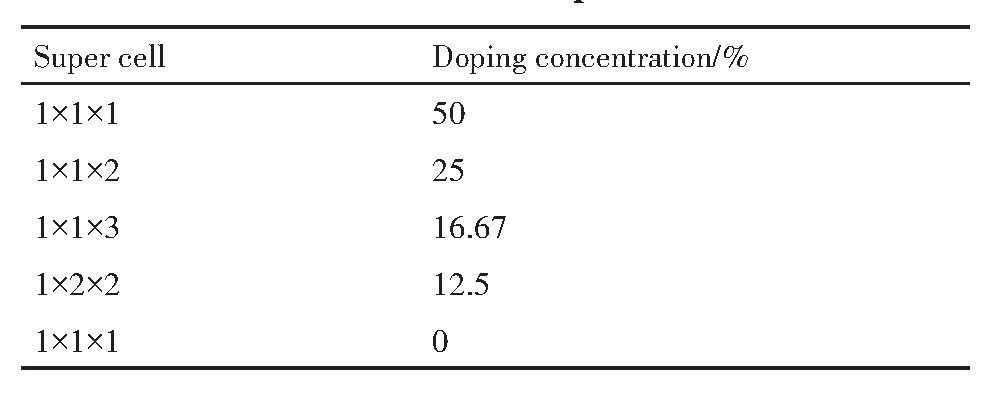
����Ľ��ʯ�ͽṹ����136 P4/MNM�ռ�Ⱥ��������ÿ��Sn O2 �����а���2��Snԭ�Ӻ�4��Oԭ�ӣ�Snԭ���ھ���Ķ�������ģ���Sn O2 ԭ���ṹ��ͼ1���趨5��La����Ũ�ȣ��ֱ�Ϊ0%,12.5%,16.67%,25%,50%����������Ũ���볬������Ӧ��ϵ���1��ʾ��
1.2���㷽��
���ľ������Ż��������ļ�������Materi�\als Studio�����е�CASTEPģ����еģ������ܶȷ������ۣ�DFT���ĵ��ռ�Norm-conserving���Ʒ��������������������ù����ݶȽ��Ʒ�����GGA
[10 ]
+PW91),GGA���Ʒ����˷���������ʵ��ϵ���ܶȱ仯���ҵ�����µ�ȱ�ݣ�����˽�������ܵļ��������Ӷ�������ܶȷ�����������ľ��ȡ��۵��Ӻ�о̬���ӵ�����ò���ͶӰ��ƽ�沨��PAW������������
[11 ,12 ]
������Ԩ������5��5��8��Monkorst-Park�����������Ż�����BFGS�㷨����Ӧ�����ֵ�趨Ϊ0.05 GPa���趨ƽ�沨�ض���Ϊ750 e V��ԭ�Ӽ�������������趨Ϊ0.3 e V?nm-1 ��������CASTEPģ���Sn O2 ������м��νṹ�Ż�ʹ��ﵽ�ȶ��ṹ��Ȼ��Ծ���������ס�����̬�ܶȺ�����ѧ���ʽ��м��㡣
ͼ1 Sn O2ԭ���ṹ
Fig.1 Model of primitive cell of Sn O2
��1 ����Ũ���볬������Ӧ��ϵ ����ԭͼ
Table 1 Correspondence relationship between doping concentration and the super cell
2�����������
2.1������ϵ�γɵ�������
�ڱȽϲ�ͬ����Ũ�ȶ�Sn O2 ������Ӱ��֮ǰ������Ӧ�Բ��ӽṹ�γɵ������Խ������ۣ���Ϊ�������ʵ�����Ƿ��ܻ����Ӧ�IJ�����Ʒ����ˣ����ĶԸ�����ģ�͵��γ��ܣ�E�� �������˼��㣬��ʽ����
[13 ]
:
ʽ�У�2 ��ϵ����������ELa ��ESn ��ʾLa,Sn��ԭ�ӻ�̬������n,m��ʾΪ���ӵ�Laԭ�������뱻�滻��Snԭ��������ʽ��1��������������ָ����ǰ��ԭ���ɵ���״̬�γɾ���ʱ���ͷŵ����������������������γɵ����׳̶ȣ����γ���Ϊ��ֵʱ�������ֵԽ��˵���˾���Խ���ϳɡ���Ϊ��ֵ����˵�������γ�
[14 ]
��
������ԭ�ӵ��������ڱ�2����ģ����Ҫ���������ļ��������ڱ�3�����ʽ��1���õ�4�ֲ�ͬ����Ũ�Ⱦ�����γ��ܣ��ɿ��������Ϊ��ֵ�����в���Ũ��Ϊ25%ʱ�����γ��ܵľ���ֵΪ���Ҳ����˵�����γ�������ǿ��
��2 ����ԭ�ӵ����� ����ԭͼ
Table 2 Energy of an isolated atom
2.2 Sn O2���������ʷ���
2.2.1��������
���������ֵ��Ǿ����ڲ��ĸ�����ģʽ����������Ծ����ȶ��Ե��о�������Ҫ�����塣�����κ�һ���ض��ṹ��������ѧҪ�����м�����ģ��Ƶ�ʶ���һ���ȵ�ʵ��ֵ�����ڲ���Ԩ������һ��������Ƶ�ʲ���Ϊ�����������������ȶ���
[15 ]
��һ����ϵ��������Ƶ���ӣ������ڸ���ģʽ�£����Ӳ��ܴ���ϵͳ��������Сֵ���������������ľ���ṹ��ȡ��������ƻ���ԭ�о���ṹ�ĶԳ��ԣ����ջᷢ��ϵͳ�Ľṹ��䡣������Ϊ����Ƶ�������㣬��ζ���������������Ӧ�ĵ��Իָ���Ҳ��Ϊ�㣬�������������������ӵ���ģʽ�����������λ�ƣ���û�лָ���ʹ���ӻص�ԭʼλ�ã������ϵ���ȶ���
Ϊ�˽�һ������Sn O2 �����ڲ�ͬLa����Ũ���µ��½ṹ�仯��ԭ��ͨ�����㴿Sn O2 �����ڲ�ͬLa����Ũ�ȵľ������ף����Է��ֲ�ͬ����Ũ�Ȼ�ı����ӵ�Ƶ�ʣ����������������Ƶ��������ýṹ�����ȶ���ͼ2(a��e������5�ֲ���Ũ�������ļ���������ͼ�����ǿ��Կ�����Ũ��N=12.5%,16.67%,50%�������׳�����Ƶ�������ýṹ�γɺ��Dz����ȶ����ڵġ�N=25%ʱ������Ԩ����һ��ʸ��Ƶ�ʶ���Ϊ������˵������������ȶ��ԡ�Ҳ����˵����Ũ��Ϊ25%�ľ���ṹ����������Ũ�ȵľ����ȶ���
2.2.2����̬�ܶȼ��ֲ�̬�ܶȷ���
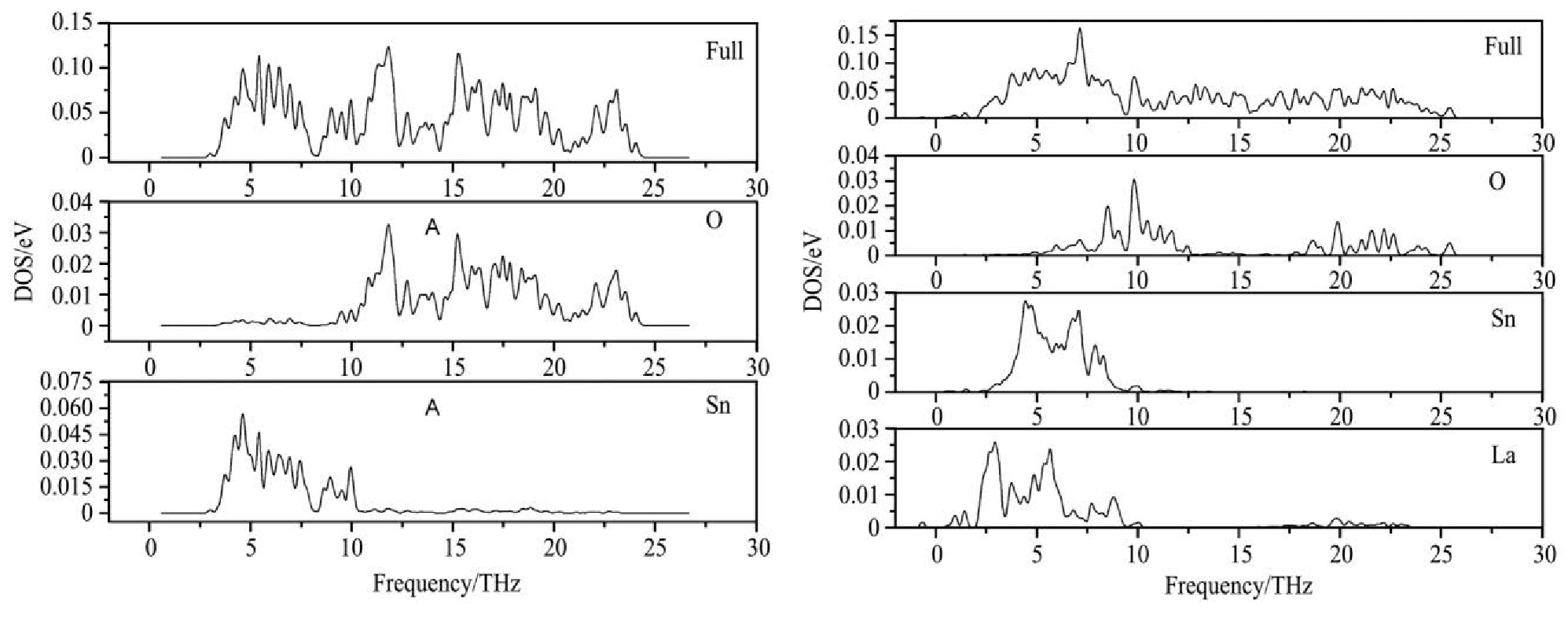
�Բ���Ũ��Ϊ0%��25%�ľ���Ϊ������������̬�ܶȼ��ֲ�̬�ܶȷ��֣�ͼ3(a,b)),Sn O2 :La(25%����������ģʽ��Ҫ������1.3��26.2 THzƵ�Σ����м�����������7.1 THz������̬�ܶȴﵽ��ֵ�����⣬������������������в�ͬ����ԭ�Ӷ�̬�ܶȵĹ��ף��������֣���ԭ�ӵķ�̬�ܶ���Ҫ�ֲ���2.5��10.0 THz,16��26 THz֮�䡣Snԭ����Laԭ�ӷֲ�̬�ܶȱȽϽӽ����ֲ���0��10 THz֮�䣻ͨ���ʹ�Sn O2 ��������̬�ܶ���Ƚϣ����Կ���Laԭ�ӵIJ��ӣ���Ҫ�Ǹı���Oԭ�ӵ���ģʽ��ʹ��Oԭ���ڵ�Ƶ�ε���ģʽ��ǿ���ڸ�Ƶ�ε���ģʽ���ͣ�������LaԪ����OԪ��֮����к�ǿ��������á�
2.3 Sn O2��������ѧ���ʷ���
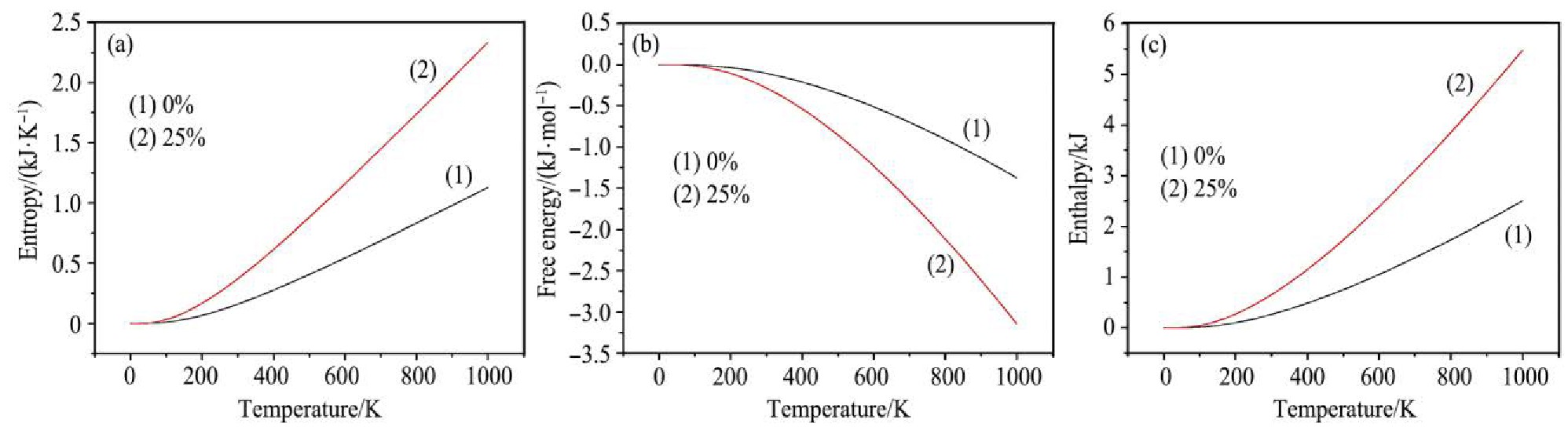
�ء��ʡ��������Ƿ�ӳ��������ѧ���ܵ���Ҫ��������г�����ƵĻ����ϣ�ֻ�Բ���Ũ��Ϊ0%��25%�ľ���Ϊ�������õõ����������Եõ����������ܡ����ӵ����Լ����ӵ��ʣ���ͼ4��ʾ��ͼ4(a���������¶����߶��ʵ����������ƣ�����Ϊ�¶����ߣ���ϵ��ԭ�ӵ��˶�Խ���ң��ɴ˰�������ϵ����������ӣ�����ϵͳ�ڷ������˶������Եı��
[16 ]
������¶����ߣ���Ҳ�����ӡ�ͼ4(b�����������������¶����߶����͵����ߣ��併���Ǿ���������������F)
[17 ]
���乫ʽΪ
[18 ]
:
��3 ��ģ����Ҫ���������ļ����� ����ԭͼ
Table 3 Calculation results of main physical parameters of each model
ͼ2 ����ɫɢͼ
Fig.2 Diagram of phonon dispersion
(a)Pure Sn O2 ;(b)Sn O2 :La(12.5%);(c)Sn O2 :La(16.67%);(d)Sn O2 :La(25%);(e)Sn O2 :La(50%)
ͼ3 ������̬�ܶȺͷֲ�̬�ܶ�ͼ
Fig.3 Diagram of total state density and partial wave state density of crystal
(a)Pure Sn O2 ;(b)Sn O2 :La(25%)
ʽ�У�U* Ϊ��ϵ���ܣ�TΪ��ϵ�¶ȣ�SΪ�ء����¶�����ʱ����ϵ�������ӣ�����ϵ����Ҳ�����ӣ�����TS���ӵ��ٶ�Ҫ����U* ���ӵ��ٶȣ����Ծ�������������¶����߳ʽ������ơ�ͼ4(c���������¶�֮��Ĺ�ϵ���ɼ���Sn O2 ������������¶����߶����ӣ��乫ʽΪ��
ͼ4 Sn O2��������ѧ�������¶ȱ仯����
Fig.4 Thermodynamic function of Sn O2 crystal varies with temperature
(a)Entropy;(b)Free energy;(c)Enthalpy
ʽ�У�PΪ��ϵѹǿ��VΪ��ϵ������ʵ�������������ϵ�ڵ�ѹ�����£��ʵ��ھ������յ���������ʽ��3����֪�����¶Ȳ������ӣ���ϵ�����������ӣ��Ҿ��巢�������ͣ�PV�����ӣ���ˣ�Sn O2 ����������¶ȵ����߶����ӡ�
����ͼ4���Է��֣�LaԪ�ز��Ӻ�ʹSn O2 ������ء������ܡ������߱�ö��͡�˵��ͬ���������벻������ϵ��ȣ���ϵ�Ļ��Ҷ����ӣ����������ܽ��ͣ����յ��������ӣ��ɴ�˵���˲��Ӻ�ľ���ϲ����ӵľ���������γɣ������ж�Ag S�\n O2 ��ͷ�����ڸ��»����²���LaԪ�غ���Ļָ�����Ҫ����δ���Ӿ���Ļָ�������
2.4���ݼ��ȵ��ʷ���
2.4.1���ݷ���
�������ӷֲ�̬�ܶȼ�����Sn O2 �����������¶ȱ仯�����ߣ���ͼ5��ʾ�����Կ�������0��300 K��Χ�ڣ�����ǰ���Sn O2 �������ݼ���������������300��700 K֮�䣬���ټ�����700 K�Ժ���������ˮƽ״̬������������һ����ֵ������Ϊ���ݵı仯����ӵ��˶�״̬��ء���ʼʱ�������¶ȵ����ߣ������˶��ķ������࣬������֮���ӣ����¶�������һ���̶�ʱ�������˶��ķ������ڱ��ͣ������ݱ㲻�����¶����߶����ӣ������ڶ�ֵ
[19 ]
���ɴˣ�����˵��0��300 K�¶ȷ�Χ�ڣ����������Ͽ죬����300 K������������û�����������ڱ��͡�
�ԱȲ���ǰ�������ݵ�������Կ�������ͬһ�¶��£����Ӻ�����������Դ���δ���Ӿ�������ݣ��ҵ���ʱ�仯�ٶȸ��졣��˵��LaԪ�صIJ��ӣ�ʹ�÷��ӵĻ�����ǿ�����μ��˶��ķ������ࡣͬʱ��ϱ������ӷֲ�̬�ܶ�ͼ��Ҳ˵����LaԪ�صIJ��Ӽ���ļ�����OԪ�صĻ��ԣ����������˾�������ݡ�
2.4.2�ȵ��ʷ���
������ȵ�����Ҫȡ�������ӵ�ƽ�����ɳ�l�͵�λ���������CV ���¶�֮��Ĺ�ϵ�������ʽΪ
[20 ]
:
����
ͼ5 �����������¶ȵĹ�ϵ
Fig.5 Relation between phonon heat capacity and temperature
3����
ͨ��Materils Studio�����е�CASTEPģ��Դ�Sn O2 ������LaԪ�صľ����������ѧ���ܵļ��㣬�õ����½��ۣ�
1.ͨ������������������õ���������ϵ���γ��ܣ��жϳ���������ϵ�γɵ������ԣ�����25%�ľ����γ��ܵľ���ֵ���
2.������������ϵ���������ף�����Ũ��Ϊ25%��������δ������Ƶ���������ȶ����ڡ������ӷֲ�̬�ܶ�֪��LaԪ����OԪ��֮�����ǿ������á�
3.���Ӻ���ϵ�Ļ��Ҷ����ӣ����������ܽ��ͣ����յ��������ӣ�˵�����Ӻ�ľ���������γɣ����ָ�����ǿ��
4.���Ӻ�����������Դ���δ���Ӿ�������ݣ�����ΪLaԪ�صIJ��Ӽ���ļ�����OԪ�صĻ��ԣ����������˾�������ݣ��Ҳ��ӻ�Ծ����ȵ��ʲ�������Ӱ�졣
5.�����������������Materials Studio�������з�����㣬��ʵ��ͨ����ͷ�����Ʊ�������������ʵ���о����õ�ʵ�����ͷ����������һ�£�������֤������㹤����ȷ�ԡ�
�����
[1] Wang J Q,Zhu Y C,Wang H T,Zhao J. Influence of additive Bi on AgSnO2 contact materials[J]. Transactions of China Electrotechnical Society, 2011, 26(1):29.�������ۣ����ʣ������Σ��Ծ�.������Bi��AgSnO2��ͷ���ϵ�Ӱ��[J].�繤����ѧ����2011,26(1):29.)
[2] Liu Z Y. Calculation and Research of Nano-Doped AgSnO2 Electrical Contact Materials[D]. Tianjin:Tianjin University,2007. 20.����־�£����ײ���AgSnO2 ��Ӵ����ϵļ������о�[D].�������ѧ��2007. 20.)
[3] Wu C P,Yi D Q,Xu C H,Zhou Y M,Weng W. Research status and development trend of silver-based alloys[J]. Electrical Materials,2012,2:1.���ⴺƼ�����࣬���ӻԣ���������Φ�������Ͻ��о���״�뷢չ����[J].�繤���ϣ�2012,2:1.)
[4] Liu Y,Xiao Z,Li Z,Li Y P,Lei Q. Properties and microstructure evolution of Cu-Cr-Ag alloy with thermomechanical treatments[J]. Chinese Journal of Rare Metals,2018,42(4):337.�����£�Ф�������ܣ�����Ƽ����ǰ.�α��ȴ�����Cu-Cr-Ag�Ͻ���֯�����ܵ�Ӱ��[J].ϡ�н�����2018,42(4):337.)
[5] Zhang X J,Wang A X,Gao B,Chen C L. Study on the phonon spectrum and vibration properties of metal ruthenium[J]. Materials Review,2015,29(4):136.���������������飬�߱����³���.�����ȵ������������ʵ��о�[J].���ϵ�����2015,29(4):136.)
[6] Dove M T. Introduction to Lattice Dynamics[M]. Cambridge:Cambridge University Press,1993. 75.
[7] Jiang W C,Chen H,Zhang W B. Study on the first principle of phonon spectrum and specific heat capacity of TATB crystal[J]. Journal of Physics,2016,65(12):220.�����IJӣ��»�����ΰ��. TATB���������������ݵĵ�һ��ԭ���о�[J].����ѧ����2016,65(12):220.)
[8] Wang Y. Development and Application of New Theoretical Methods for Complex Molecular Systems[D].Shanghai:East China Normal University,2018. 15.����ӱ.���ӷ�����ϵ�������·����ķ�չ��Ӧ��[D].�Ϻ�������ʦ����ѧ��2018. 15.)
[9] Mu X. First-Principles Study of Electron Spin Polarization of Some Materials[D]. Hefei:University of Science and Technology of China,2011. 17.������.��һ��ԭ���о�һЩ���ϵĵ�����������[D].�Ϸʣ��й���ѧ������ѧ��2011. 17.)
[10] Liu X J,Yang J C,Jia G X,Yang C Q,Cai C K. A first principle study on occupancy and electronic structure of V,Mn,Mg in��-Fe((N)[J]. Chinese Journal of Rare Metals,2019,43(12):1357.���������������ֹ���������ţ��̳��j. V,Mn,Mg�ڨ�-Fe(N����ռλ�����ӽṹ�ĵ�һ��ԭ���о�[J].ϡ�н�����2019,43(12):1357.)
[11] Torrent M,Jollet F,Bottin F. Implementation of the projector augmented-wave method in the ABINIT code:application to the study of iron under pressure[J]. Comput. Mater. Sci.,2008,42(2):337.
[12] Kresse G,Joubert D. From ultra soft pseudo potentials to the projector augmented-wave method[J]. Phys.Rev. B,1999,59(3):1758.
[13] Cui X Y,Medvedeva J E,Delley B,Freeman A J,Newan N. Role of embedded clustering in dilute magnetic semiconductorsn:Cr doped GaN[J]. Phys. Rev.Lett.,2005,95(25):6404.
[14] Ghosh G,Asta M. First-principles calculation of structural energetics of Al-TM(TM=Ti,Zr,Hf)intermetallics[J]. Acta Materialia,2005,53(11):3225.
[15] Born M,Huang K. Dynamical Theory of Crystal Lattices[M]. Oxford:Oxford University Press,1998. 73.
[16] Su H,Gao X,Hu B W,Yuan Z X,Li W X. Study on entropy change law based on particle discrete element rock model[J]. International Journal of Hydroelectric Energy,2018,36(6):106.���ԣ������������ģ�Ԭ���飬������.���ڿ�����ɢԪ��ʯģ�͵��ر�����о�[J].ˮ����Դ��ѧ��2018,36(6):106.)
[17] Xu W. Preparation and Growth Mechanism of Zinc Oxide Crystal Materials[D]. Xi'an:Xi'an University of Science and Technology,2008. 16.������.����п������ϵ��Ʊ��������������о�[D].�����������Ƽ���ѧ��2008. 16.)
[18] He C Y. Phase Diagram Determination and Thermodynamic Study of Al-Cu-Mn,Al-Cu-Si,Al-Mg-Ni and NiTi-Si Systems[D]. Changsha:Central South University,2008. 13.���ش���. Al-Cu-Mn��Al-Cu-Si��Al-Mg-Ni��Ni-Ti-Si��ϵ����ͼ�ⶨ������ѧ�о�[D].��ɳ�����ϴ�ѧ��2008. 13.)
[19] He W Z,Fang Z J. First-principles study of phonon spectrum and thermodynamic properties of LiNiPO4 [J]. Journal of Guangxi University of Science and Technology,2014,25(4):14.�������У���־��. LiNiPO4 ����������ѧ���ʵ�һ��ԭ���о�[J].�����Ƽ���ѧѧ����2014,25(4):14.)
[20] Cao Q X,Lei T M,Huang Y X. The Basis of Solid Physics[M]. Xian:Xi'an University of Electronic Science and Technology Press,2008. 101.����ȫϲ��������������ϼ.��������ѧ����[M].�������������ӿƼ���ѧ�����磬2008. 101.)