DOI: 10.11817/j.ysxb.1004.0609.2020-35802
合成条件对电解锰渣制备水化硅酸钙过程中结构和性能的影响
李昌新1,喻 源1,张庆武1,汪 洋1,钟 宏2, 3,王 帅2, 3
(1. 南京工业大学 安全科学与工程学院,南京211816;
2. 中南大学 化学化工学院,长沙410083;
3. 中南大学 锰资源高效清洁利用湖南省重点实验室,长沙 410083)
摘 要:基于电解锰渣成分及水化硅酸钙(C-S-H)材料的结构特性,提出以电解锰渣为原料制备C-S-H材料,开发电解锰渣基C-S-H材料制备新技术。系统研究由电解锰渣制备C-S-H材料过程中反应pH值、反应温度、晶化时间等因素对合成C-S-H材料的矿相、微观结构和溶钙性能的影响。结果表明:C-S-H产品的种类、微观结构及其溶钙性能与反应工艺条件紧密相关,在优化工艺条件下(反应pH值12.0、反应温度100 ℃、晶化时间10 h)制备所得的C-S-H材料结构酥松,其比表面积为205.0 m2/g,总孔孔容为0.68 cm3/g,且溶钙能力最强,溶出钙离子浓度为11.52 mg/L,适合作为吸附除磷材料在水处理过程中使用。
关键词:电解锰渣;水化硅酸钙;合成条件;微观结构;溶钙性能
文章编号:1004-0609(2020)-06-1368-09 中图分类号:X757;TD981 文献标志码:A
我国电解锰工业在迅速发展的同时,也引发了严重的环境污染问题,其中锰渣污染尤为突出。锰渣的大量堆存必然占用大片的土地,增加了企业生产成本,制约了行业的可持续发展,同时,锰渣长时间暴露于自然环境下会使得其中一部分有害物质进入土壤、水体中,进而对生态系统造成危害,锰渣中的重金属对土壤和水环境的影响已突出显现[1-2]。因此,如何有效地利用电解锰渣成了当今亟待解决的问题。
近年来,针对电解锰渣资源化的问题,不少学者进行了有益探索,设计发展出了以下利用途径:金属元素提取回收工艺[3-4]、建筑材料制备工艺[5]、陶瓷材料制备工艺[6]、锰渣复合肥制备工艺[7]、水泥材料制备工艺[8]等。虽然一些研究项目的应用前景看好,但受到产品产量、附加值、工艺技术条件和成本的限制,工业化利用方面进展缓慢,在实际工业生产中存在锰渣利用效率低、工艺复杂、成本高、经济效益差、环境风险高等问题,严重制约了资源综合利用水平的提升。因此,迫切需要不断研发新型、高效的锰渣资源化技术以解决闲置堆存锰渣的开发利用问题。
基于电解锰渣成分及水化硅酸钙(C-S-H)材料的结构特性,本文提出以电解锰渣为原料制备C-S-H材料,开发电解锰渣基C-S-H材料制备新技术。同时,C-S-H材料作为一种以生石灰、石英粉为原料经水热合成的无机材料[9],该材料一方面能够自发溶解Ca2+和OH-[10-12],溶出的Ca2+、OH-与废水中的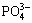 在其表面结晶,逐渐转化为羟基磷灰石,实现磷资源的回收[13],另一方面,C-S-H材料作为阳离子交换剂,可以将重金属及放射性元素稳定固化在其晶格内部[14-15],因此,C-S-H材料适合作为水环境净化吸附材料广泛使用。总之,以电解锰渣为原料制备C-S-H材料,不仅可以实现电解锰渣的资源化利用,同时也符合矿业行业绿色发展的理念和要求。
在其表面结晶,逐渐转化为羟基磷灰石,实现磷资源的回收[13],另一方面,C-S-H材料作为阳离子交换剂,可以将重金属及放射性元素稳定固化在其晶格内部[14-15],因此,C-S-H材料适合作为水环境净化吸附材料广泛使用。总之,以电解锰渣为原料制备C-S-H材料,不仅可以实现电解锰渣的资源化利用,同时也符合矿业行业绿色发展的理念和要求。
本文作者在前期的研究中,成功地实现了电解锰渣基C-S-H材料的制备[16],但是,关于不同合成条件对C-S-H生成的影响尚缺乏系统性研究,本文作者以电解锰渣为原料,通过水热合成法制备C-S-H材料,研究反应体系中反应pH值、反应温度、晶化时间等合成条件对C-S-H材料的矿相、微观结构和溶钙性能的影响,优化C-S-H材料制备过程的工艺条件,对电解锰渣的有效利用开拓新的途径。
1 实验
1.1 材料
本实验所用锰渣为湘西某电解锰厂渣库中堆存的废渣,pH为5.50,水溶性物质占24.43%(质量分数)。锰渣中主要化学成分(质量分数)如下:SiO2(24.6%),Al2O3(12.25%),MnO(4.6%),Fe2O3(7.9%),CaO(8.6%),锰渣的物相构成非常的复杂,但锰渣的物相构成主要为:石英(SiO2),石膏(CaSO4・2H2O)。试验用硅酸钠、氢氧化钠、PEG 400、无水乙醇为分析纯,水为去离子水。
1.2 实验方法
电解锰渣经2 mol/L HNO3于80 ℃下活化2 h后,过滤得到浸出渣及浸出液,其中浸出渣的综合利用见本课题组的前期研究,锰渣基C-S-H(C-S-H)的制备以浸出液为原料。C-S-H的制备工艺如图1所示。本研究中,C-S-H的制备采用碱硅酸盐与钙盐的溶液反应法,根据前期工作及文献报道,选取n(Ca)/n(Si)=1:1,在制备C-S-H时首先将一定体积的2#滤液逐滴滴加到100 mL 0.1 mol/L硅酸钠溶液中,同时保证搅拌均匀,其中硅酸钠溶液中先预溶0.6 mL/L的PEG 400;待全部滴加完全时以0.5 mol/L氢氧化钠溶液调整溶液至一定pH值,然后将上述悬浮液于一定温度下剧烈搅拌反应2 h后,继续在该温度下晶化一定时间。晶化完成后过滤、先后用去离子水和无水乙醇充分洗涤、过滤并于80 ℃干燥4 h得到C-S-H粉体。由于,反应体系中反应pH值、反应温度、晶化时间等与C-S-H产品的晶型、溶钙供碱性能等性能直接相关。因此,本文系统地研究了反应体系中反应pH值、反应温度、晶化时间等对产品质量的影响。
1.3 测试分析方法
固体物质化学组成采用荷兰PANalytical公司Axios型X射线荧光光谱仪(XRF)测定;溶液中各物质的化学组成采用美国Perkin EImer公司Optima5300型电感耦合等离子体原子发射光谱(ICP)测定;采用德国Bruker公司D8 Discover 2500 X-射线粉末衍射仪测定锰渣及C-S-H产品的物相组成;采用捷克TESCAN公司Mira3型扫描电子显微镜进行产品微观形貌分析;产物骨架结构采用美国Nicolet公司Nicolet 6700型傅里叶变换红外光谱仪测定(KBr压片);C-S-H溶钙性能的测定方法如下:向装有100 mL蒸馏水的250 mL锥形瓶中加入所制备得到的C-S-H 0.4 g在25 ℃、100 r/min恒温恒速搅拌条件下,在预设的时间间隔下,取上层清液,离心后测定溶液中Ca2+的浓度。
2 结果和讨论
2.1 反应pH值的影响

图1 电解锰渣制备C-S-H实验流程图
Fig. 1 Schematic diagram of synthetic routes from EMR to C-S-H
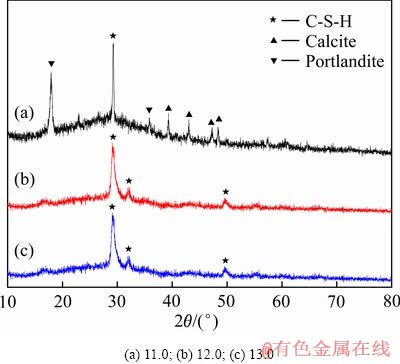
图2 不同反应pH值下C-S-H的XRD谱
Fig. 2 XRD patterns of synthesized C-S-H under various pH value
图2所示为不同反应pH值在初始n(Ca)/n(Si)=l.0、反应温度100 ℃、晶化10 h的条件下所制备C-S-H的XRD谱。如图2所示,不同反应pH值下所制备得到的C-S-H产物的XRD谱有着比较明显的差异。当反应pH值在11.0时,主要产物为C-S-H及少量的CaCO3和Ca(OH)2。这是因为C-S-H是一种高度复杂的体系,其基本组成为SiO2-CaO-H2O,在大多数情况下C-S-H中都混杂了部分Ca(OH)2,当反应体系的碱度较低时, Ca(OH)2和[SiO4]4-四面体不能很好的以“键合质”形式结合,而是游离出来。这一现象与C-S-H的“固溶模型”一致[17]。C-S-H的“固溶模型”认为其是托贝莫来石和Ca(OH)2的共溶体,Ca(OH)2被吸附在托贝莫来石的结构层间。而CaCO3的生成主要是由于存在于C-S-H纳米颗粒间的Ca(OH)2与溶于反应体系中的CO2反应所致。当反应pH值大于12.0时,反应产物在2θ分别为16.6°、29.2°、31.9°、49.5°、54.9°的位置上呈现明显的衍射峰,以上衍射峰位与相关文献上报道的纯C-S-H的XRD特征峰基本一致[18-19],说明在高碱度的条件下产物为纯C-S-H凝胶。同时图2表明,随着反应pH值的增加,样品的特征峰愈加尖锐,C-S-H结构的有序程度越来越高,可见反应pH值对合成的C-S-H凝胶的结构有一定的影响。这可能是随着碱度的提高,C-S-H凝胶和Ca(OH)2的共溶体结合更加稳定,有利于提高产物有序度和结晶度。
图3所示为不同反应pH值在初始n(Ca)/n(Si)=l.0、反应温度100 ℃、晶化10 h的条件下所制备C-S-H的SEM像。由图3可知,随着反应pH值的增加,产物的结构发生较大变化,当反应pH值为11.0时,产物主要由细小的颗粒状物质组成,当反应pH值增加至12.0时,产品主要由弯曲的纤维状物质组成,纤维状物质互相缠绕链接整体形成网络状,孔洞较均匀,纤维之间缠绕较紧密,但存在细小的孔洞。当反应pH值继续增加至13.0后,形貌与反应pH值为12.0时相近,从形态上看样品结构更加致密,表面出现了大量的絮状小块体碎屑颗粒。

图3 不同反应pH值下C-S-H的SEM像
Fig. 3 SEM images of synthesized C-S-H under various pH value
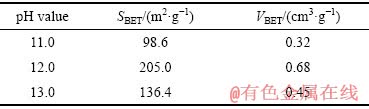
采用氮吸附法(BET)分别对不同反应pH值在初始n(Ca)/n(Si)=l.0,反应温度100 ℃,晶化10 h的条件下所制备C-S-H粉体的比表面积和孔容进行测定,其结果如表1所列。从表1可以看出,反应pH值能显著影响C-S-H材料的比表面积和孔容积,随着反应pH值从11.0增至13.0时,C-S-H材料的孔容及比表面积均先增加后减小,且C-S-H粉体比表面积较大,最大可达205 m2/g,以上结果与前述SEM像的分析结果基本吻合。
表1 不同反应pH值下C-S-H材料的总孔孔容及比表面积
Table 1 BET pore volume and specific surface area of C-S-H synthesized under various pH values
由于[SiO4]4-四面体所处位置不同,C-S-H中各个化学键具有不同的特征振动波数,与XRD相比,FT-IR能从基团和原子水平显示物质的结构,因此在本文中除了采用XRD,SEM分析外,并以傅里叶红外仪对样品的结构做了详细的研究。不同反应pH值在初始n(Ca)/n(Si)=l.0,反应温度100 ℃,晶化10 h的条件下所制备C-S-H的红外图谱如图4所示。根据YU等[20]的研究,C-S-H具有和1.4 nm托勃莫来石相似的结构,并且在波数970 cm-1附近存在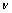 (Si―O) Q2伸缩振动特征吸收峰。从图4中可以看出,随着反应pH值增加,该吸收峰位置向低波数移动,这与文献报道结果基本一致。分析认为此吸收峰的红移是由于在高pH条件下,Ca2+离子与非桥氧(Si―O-)结合以形成Si―O―Ca―O―Si链。在不同pH值下所得样品的FT-IR图谱中均发现,450 cm-1波数附近的δ(Si―O―Si)弯曲振动,660 cm-1波数附近的
(Si―O) Q2伸缩振动特征吸收峰。从图4中可以看出,随着反应pH值增加,该吸收峰位置向低波数移动,这与文献报道结果基本一致。分析认为此吸收峰的红移是由于在高pH条件下,Ca2+离子与非桥氧(Si―O-)结合以形成Si―O―Ca―O―Si链。在不同pH值下所得样品的FT-IR图谱中均发现,450 cm-1波数附近的δ(Si―O―Si)弯曲振动,660 cm-1波数附近的 (Si―O―Si)对称伸缩振动,1640 cm-1波数附近的δ(O―H)弯曲振动,3420 cm-1波数附近的(O―H)伸缩振动。其中,样品中O―H键可能是[SiO4]4-四面体中的O―H键,也可能是C-S-H结构中水分子中的O―H键。同时所有C-S-H样品中均在1400 cm-1波数附近处有明显的吸收峰,该峰归属于
(Si―O―Si)对称伸缩振动,1640 cm-1波数附近的δ(O―H)弯曲振动,3420 cm-1波数附近的(O―H)伸缩振动。其中,样品中O―H键可能是[SiO4]4-四面体中的O―H键,也可能是C-S-H结构中水分子中的O―H键。同时所有C-S-H样品中均在1400 cm-1波数附近处有明显的吸收峰,该峰归属于 (
( )非对称伸缩振动,因为样品一旦置于空气环境中就无法避免CO2的吸收。将上述FT-IR图谱与文献中所列的C-S-H标准红外图谱特征波数对照,可以发现在pH值12.0~13.0条件下合成的C-S-H与C-S-H标准红外图谱非常吻合[21],再次表明以2#滤液为钙源的C-S-H的成功制备。
)非对称伸缩振动,因为样品一旦置于空气环境中就无法避免CO2的吸收。将上述FT-IR图谱与文献中所列的C-S-H标准红外图谱特征波数对照,可以发现在pH值12.0~13.0条件下合成的C-S-H与C-S-H标准红外图谱非常吻合[21],再次表明以2#滤液为钙源的C-S-H的成功制备。
含钙材料能够实现除磷的关键在于其具有溶钙供碱的特性,因此考察所制备的C-S-H的溶钙能力是十分必要的。于各种反应pH值下制备的C-S-H的溶钙曲线如图5所示。由图5可知,当达到反应平衡时,于pH 11.0、12.0、13.0下制备的C-S-H溶出钙浓度分别为8.64、11.52、10.02 mg/L。由此可见,当反应pH在12.0时,C-S-H的溶钙性能较好。探究其原因认为,当反应pH值为11.0时,生成了部分的CaCO3而其溶钙能力不如C-S-H,而在反应pH值为13.0时,由于碱度进一步增加,C-S-H类物质晶相生长得越完整,结构就越紧密,比表面积降低,对材料的孔结构形成阻塞,阻碍了材料内部Ca2+的持续溶出。
基于上述研究表明,在合成制备C-S-H凝胶过程中选择合适的反应pH值对产品的纯度、微观结构、溶钙供碱能力是至关重要的。在本研究中,选择反应pH值为12.0是合适的。

图4 不同反应pH值下C-S-H红外图谱
Fig. 4 FT-IR spectra of synthesized C-S-H under various pH value

图5 反应pH值对C-S-H溶钙能力影响
Fig. 5 Concentration of Ca2+ dissolved by synthesized C-S-H under various pH value
2.2 反应温度的影响
在初始n(Ca)/n(Si)=l.0,反应pH值12.0,晶化时间10 h的条件下考察了反应温度对制备C-S-H的影响,并以XRD、SEM、FT-IR、溶钙供碱能力等方法进行了评价。图6所示为在不同反应温度条件下所制备C-S-H的XRD谱。从图6中可以看出,在以上3种不同的反应温度下所制备C-S-H XRD谱中均含有C-S-H的衍射峰,说明在反应温度为60 ℃时便开始生成C-S-H。随着反应温度的升高,C-S-H的衍射主峰(d=0.307 nm)逐渐增强,说明随着反应温度升高,C-S-H样品的纯度逐步增加。而当反应温度在60 ℃时,样品中存在CaCO3、Ca(OH)2、SiO2。这是由于当反应温度较低时,空气中的CO2更容易进入反应体系,从而产生CaCO3。相比反应温度为80 ℃时,样品中CaCO3、SiO2均消失,同时Ca(OH)2的峰强也大幅下降,也就是说随着反应温度的增加,反应体系中Ca(OH)2慢慢从开始的游离状态逐渐融入C-S-H结构中,符合水化硅酸钙的“固溶模型”。

图6 不同反应温度下水化硅酸钙XRD谱
Fig. 6 XRD patterns of synthesized C-S-H under various reaction temperatures
图7所示为不同反应pH值在初始n(Ca)/n(Si)=l.0,反应pH值12.0,晶化10 h的条件下所制备C-S-H的SEM像。由图7所示,不同反应温度下制备的C-S-H微观结构有较大差异,随着反应温度的上升,粉末从开始比较密实的颗粒(60 ℃)变化为表面相对疏松的团状颗粒(80 ℃),随着反应温度的进一步增大(100 ℃),C-S-H粉体主要由弯曲的纤维状物质组成,纤维状物质互相缠绕链接整体形成网络状,此时,C-S-H结构最为酥松,材料表面分布有较多的孔隙[22]。表2所列为不同反应温度下合成的C-S-H的孔结构及比表面积,由表2所示,不同反应温度下合成所得的C-S-H的比表面积和总孔孔容差异较大,反应温度为100 ℃合成的C-S-H比表面积最大,达到205.0 m2/g,总孔孔容为0.68 cm3/g,而反应温度为60 ℃的C-S-H比表面积最小,仅为58.2 m2/g,总孔孔容为0.18 cm3/g。
图8所示为在不同反应温度下所制备C-S-H的红外图谱。由图8可知,在不同反应温度的C-S-H样品红外图谱振动峰位置基本一致。随着反应温度从60 ℃增加到100 ℃时,各个样品的红外特征峰中位于波数970 cm-1附近的(Si―O) Q2伸缩振动变化最为明显,其吸收峰波数从1024 cm-1红移至973 cm-1。分析其原因认为,当反应温度升高,Ca(OH)2反应活性增加,更多的Ca2+进入到C-S-H体系中促进了Si―O―Ca键大量形成。

图7 不同反应温度下C-S-H的SEM像
Fig. 7 SEM images of synthesized C-S-H under various reaction temperatures
表2 不同反应温度下C-S-H材料的总孔孔容及比表面积
Table 2 BET pore volume and specific surface area of C-S-H synthesized under various reaction temperature

图9所示为在不同反应温度下制备所得的C-S-H溶钙曲线。由图9可知,不同反应温度合成的C-S-H材料在水中Ca2+溶出浓度存在差异,其中,当反应温度为100 ℃时,溶出Ca2+浓度达到11.52 mg/L,而反应温度为60 ℃时,溶出Ca2+浓度仅为7.14 mg/L。结合在不同反应温度下制备C-S-H产品性状,于100 ℃下制备的C-S-H为粉状,而在低温下制备的C-S-H略成块状,表明C-S-H材料溶钙能力与其微观结构有关,产品的结构愈疏松,比表面积越大,其溶钙能力越强,反之越弱。
综合以上分析,表明反应温度对产品结构、溶钙供碱能力影响显著,而溶钙能力直接与产品除磷性能相关。因此在制备C-S-H过程中控制反应温度在100 ℃较优。

图8 不同反应温度下水化硅酸钙红外图谱
Fig. 8 FT-IR spectra of synthesized C-S-H under various reaction temperatures

图9 反应温度对水化硅酸钙溶钙能力影响
Fig. 9 Concentration of Ca2+ dissolved by synthesized C-S-H under various reaction temperatures
2.3 晶化时间的影响
控制反应体系初始n(Ca)/n(Si)=l.0,反应pH值12.0,反应温度100 ℃,分别以XRD、SEM、溶钙性能等方法研究晶化时间对制备C-S-H的影响。图10所示为在不同晶化时间下所制备C-S-H的XRD图谱。由图10可知,晶化时间从10 h延长到24 h对C-S-H的晶相没有明显影响,但晶化时间对制备所得的C-S-H微观结构有较大影响,随着晶化时间的延长,制备的C-S-H变得越紧密(如图11所示),能够明显地看出弯曲的薄片状物质增多,絮状物减少,这是由于絮状的C-S-H相逐渐转化为结晶度较高的托勃莫来石及硬硅钙石。表3所列为不同晶化时间下合成的C-S-H的孔结构及比表面积,由表3所列,所制C-S-H材料随晶化时间的延长,比表面积、孔容积均逐渐减小。
图12所示为不同晶化时间下制备所得的C-S-H溶钙曲线,由图12可知,晶化时间为10 h的C-S-H溶出的Ca2+浓度最大,而晶化时间为24 h的C-S-H溶出的 Ca2+浓度最小。结合图11和表3可以发现,C-S-H材料的溶钙能力与其微观结构有密切关联,随着晶化时间增加,C-S-H晶相生长得越完整,结构越紧密,释放出钙离子的能力变差,晶化时间为10 h合成的C-S-H的孔隙结构较为疏松,且比表面积最大,因而其溶钙能力最强。
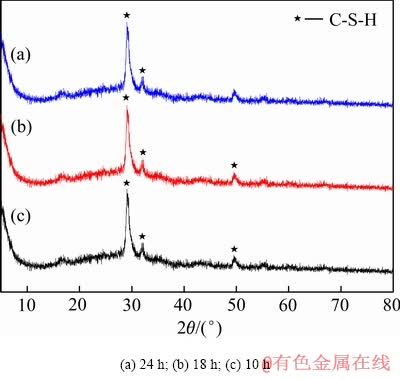
图10 不同晶化时间下水化硅酸钙XRD谱
Fig. 10 XRD patterns of synthesized C-S-H under various crystallization time

图11 不同晶化时间下C-S-H的SEM像
Fig. 11 SEM images of synthesized C-S-H under various crystallization time
表3 不同晶化时间下C-S-H材料的总孔孔容及比表面积
Table 3 BET pore volume and specific surface area of C-S-H synthesized under various crystallization time

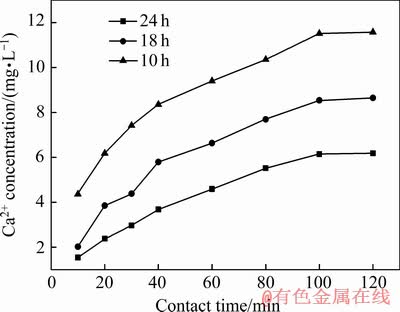
图12 晶化时间对C-S-H溶钙能力影响
Fig. 12 Concentration of Ca2+ dissolved by synthesized C-S-H under various crystallization time
3 结论
1) 采用电解锰渣为主要原料制备C-S-H材料,研究水热合成体系中反应pH值、反应温度、晶化时间等因素对合成C-S-H材料的矿相、微观结构和溶钙性能的影响。
2) 在反应pH值为12.0、反应温度为100 ℃、晶化时间为10 h的条件下制备所得的C-S-H材料结构酥松,比表面积为205.0 m2/g,总孔孔容为0.68 cm3/g,且溶钙能力最强,溶出钙离子浓度为11.52 mg/L,适合作为吸附除磷材料在水处理过程中使用。
3) 以电解锰渣为原料制备C-S-H材料,开发电解锰渣基C-S-H材料制备新技术,既可以实现电解锰渣中有用成分的综合利用,并能得到附加值高的系列化电解锰渣基C-S-H材料,有利于我国锰渣资源的高效开发利用及环境保护。
REFERENCES
[1] DUAN N, WANG F, ZHOU C B, ZHU C L, YU H B. Analysis of pollution materials generated from electrolytic manganese industries in China[J]. Resources, Conservation and Recycling, 2010, 54(8): 506-511.
[2] HU N, ZHENG J F, DING D X, LIU J, YANG L Q, YIN J, LI G Y, WANG Y D, LIU Y L. Metal pollution in Huayuan river in Hunan province in China by manganese sulphate waste residue[J]. Bulletin of Environmental Contamination and Toxicology, 2009, 83(4): 583-590.
[3] 杨晓红,向 欣, 薛希仕. 电解锰渣酸浸实验条件探究[J]. 硅酸盐通报, 2018, 37(7): 2326-2330.
YANG Xiao-hong, XIANG Xin, XUE Xi-shi. Study on acid leaching experimental conditions of electrolytic manganese residue[J]. Bulletin of the Chinese Ceramic Society, 2018, 37(7): 2326-2330.
[4] 陈红亮, 张玉涛, 张秋云, 李 琳. 酸法还原浸出电解锰渣中锰和铁的工艺条件和动力学分析[J]. 硅酸盐通报, 2017, 36(8): 2844-2849.
CHEN Hong-liang, ZHANG Yu-tao, ZHANG Qiu-yun, LI Lin. Technology conditions and kinetics analysis of manganese and iron ions leaching from electrolytic manganese residue by acid reduction[J]. Bulletin of the Chinese Ceramic Society, 2017, 36(8): 2844-2849.
[5] 佘 思, 黄海燕, 马泽宇, 杨立凡. 可用于建筑材料的电解锰渣性能试验研究[J]. 中国锰业, 2017, 35(1): 123-125.
SHE Si, HUANG Hai-yan, MA Ze-yu, YANG Li-fan. A test research of EMM slag in construction materials[J]. China's Manganese Industry, 2017, 35(1): 123-125.
[6] WU F F, LI X P, ZHONG H, WANG S. Utilization of electrolytic manganese residues in production of porous ceramics[J]. International Journal of Applied Ceramic Technology, 2016, 13(3): 511-521.
[7] TAKEHARA H, SHIMIZUGAWA R, TOKAI T. Manganese fertilizer: United States Patent, US5749935[P]. 1998-05-12.
[8] 黄 川, 史晓娟, 龚 健, 陈绍杨. 碱激发电解锰渣制备水泥掺合料[J]. 环境工程学报, 2017, 11(3): 1851-1856.
HUANG Chuan, SHI Xiao-juan, GONG Jian, CHEN Shao-yang. Alkali-activated electrolytic manganese residue preparation of cement admixture[J]. Chinese Journal of Environmental Engineering, 2017, 11(3): 1851-1856.
[9] RICHARDSON I G. The calcium silicate hydrates[J]. Cement & Concrete Research, 2008, 38(2): 137-158.
[10] RICHARDSON I G. Tobermorite/jennite-and tobermorite/ calcium hydroxide-based models for the structure of C-S-H: Applicability to hardened pastes of tricalcium silicate, β-dicalcium silicate, Portland cement, and blends of Portland cement with blast-furnace slag, metakaol or silica fume[J]. Cement & Concrete Research, 2004, 34(9): 1733-1777.
[11] LIN K L, CHANG J, WANG Z. Fabrication and the characterisation of the bioactivity and degradability of macroporous calcium silicate bioceramics in vitro[J]. Journal of Inorganic Materials, 2005, 72(8): 1341-1347.
[12] CHEN J J, THOMAS J J, TAYLOR H F W, JENNINGS H M. Solubility and structure of calcium silicate hydrate[J]. Cement & Concrete Research, 2004, 34(9): 1499-1519.
[13] 关 伟, 吉芳英, 陈晴空, 晏 鹏, 张 千. 水化硅酸钙的制备及磷回收性能表征[J]. 功能材料, 2016, 43(23): 3286-3290.
GUAN Wei, JI Fang-ying, CHEN Qing-kong, YAN Peng, ZHANG Qian. Preparation and phosphorus recovery performance of porous calcium-silicate-hydrate[J]. Journal of Functional Materials, 2016, 43(23): 3286-3290.
[14] COLEMAN N J. Interactions of Cd(Ⅱ) with waste-derived 11  tobermorites[J]. Separation & Purification Technology, 2005, 48(1): 62-70.
tobermorites[J]. Separation & Purification Technology, 2005, 48(1): 62-70.
[15] COLEMAN N J, BRASSINGTON D S, RAZA A, MENDHAM A P. Sorption of Co2+ and Sr2+ by waste- derived 11 tobermorite[J]. Waste Management, 2006, 26(3): 260-267.
[16] LI C X, ZHONG H, WANG S, XUE J R, WU F F, ZHANG Z Y. Preparation of MnO2 and calcium silicate hydrate from electrolytic manganese residue and evaluation of adsorption properties[J]. Journal of Central South University, 2015, 22(7): 2493-2502.
[17] 张文生, 王宏霞, 叶家元. 水化硅酸钙的结构及其变化[J]. 硅酸盐学报, 2005(1): 63-68.
ZHANC Wen-sheng, WANC Hong-xia, YE Jia-yuan. Structure and its variation of calcium silicate hydrates[J]. Journal of the Chinese Ceramic Society, 2005(1): 63-68.
[18] 雷永胜, 韩 涛, 王慧奇, 靳秀芝, 杨 芳, 曹红红, 程芳琴. 水热合成水化硅酸钙(C-S-H)的制备与表征[J]. 硅酸盐通报, 2014, 33(3): 465-469.
LEI Yong-sheng, HAN Tao, WANG Hui-qi, JIN Xiu-zhi, YANG Fang, CAO Hong-hong, CHENG Fang-qin. Preparation and characterization of calcium silicate hydrate (C-S-H) synthesized by the hydrothermal method[J]. Bulletin of the Chinese Ceramic Society, 2014, 33(3): 465-469.
[19] KUWAHARA Y, TAMAGAWA S, FUJITANI T, YAMASHITA H. A novel conversion process for waste slag: synthesis of calcium silicate hydrate from blast furnace slag and its application as a versatile adsorbent for water purification[J]. Journal of Materials Chemistry A, 2013, 1(24): 7199-7210.
[20] YU P, KIRKPATRICK R J, POE B, MCMILLAN P F, CONG X D. Structure of calcium silicate hydrate (C-S-H): Near-, mid-, and far-infrared spectroscopy[J]. Journal of the American Ceramic Society, 1999, 82(3): 742-748.
[21] MEISZTERICS A, ROSTA L, PETERLIK H, ROHONCZY J, KUBUKI S, HENITS P, SINKO K. Structural characterization of gel-derived calcium silicate systems[J]. The Journal of Physical Chemistry A, 2010, 114(38): 10403-10411.
[22] JOHNSTON J H, SMALL A C. Photoactivity of nano- structured calcium silicate-titanium dioxide composite materials[J]. Journal of Materials Chemistry, 2011, 21(4): 1240-1245.
Effects of synthesis conditions on formation process and property of calcium silicate hydrate prepared from electrolytic manganese residue
LI Chang-xin1, YU Yuan1, ZHANG Qing-wu1, WANG Yang1, ZHONG Hong2, 3, WANG Shuai2, 3
(1. College of safety Science and Engineering, Nanjing Technology University, Nanjing 211816, China;
2. College of Chemistry and Chemical Engineering, Central South University, Changsha 410083, China;
3. Hunan Provincial Key Laboratory of Efficient and Clean Utilization of Manganese Resources, Central South University, Changsha 410083, China)
Abstract: Based on the composition of electrolytic manganese residue (EMR) and the structural characteristics of hydrated calcium silicate (C-S-H) material, a novel technology was used for EMR based C-S-H material preparation using EMR as raw material. Calcium silicate hydrate (C-S-H) was synthesized from electrolytic manganese residue (EMR) via a hydrothermal synthetic procedure. The major factors influencing the crystalline phase, microstructure and Ca2+ release capacity of C-S-H were investigated in the aspects of reaction pH value, reaction temperature and crystallization time. The results show that the crystalline phase, microstructure and Ca2+ release capacity of C-S-H are greatly affected by the reaction pH value, reaction temperature and crystallization time during the preparation process. And the optimal condition for C-S-H synthesized from electrolytic manganese residue is determined to reaction pH value of 12.0, reaction temperature of 100 ℃ and crystallization time of 10 h. And the product synthesized under the optimal condition is identified to be a calcium silicate hydrate with a specific surface area and pore volume of 205 m2/g and 0.68 cm3/g, respectively. Meanwhile, under the above conditions, Ca2+ release capacity is 11.52 mg/L, making this material a promising candidate for removal of phosphate ions diluted in wastewater.
Key words: electrolytic manganese residue; calcium silicate hydrate; synthesis condition; microstructure; Ca2+ release capacity
Foundation item: Project(2017M611799) supported by the China Postdoctoral Science Foundation; Project(BK20190690) supported by the Basic Research Program (Natural Science Foundation)―Youth Foundation Project of Jiangsu Province, China; Project(2016TP1007) supported by the Hunan Provincial Science and Technology Plan Project, China; Project(MN2018K02) supported by the Open Research Fund of Hunan Provincial Key Laboratory of Efficient and Clean Utilization of Manganese Resources, China
Received date: 2019-04-29; Accepted date: 2019-10-28
Corresponding author: LI Chang-xin; Tel: +86-25-58139551; E-mail: lichangxin2010@njtech.edu.cn;
lichangxin20160706@163.com
(编辑 李艳红)
基金项目:中国博士后科学基金资助项目(2017M611799);江苏省基础研究计划(自然科学基金)―青年基金项目(BK20190690);湖南省科技计划项目(2016TP1007);锰资源高效清洁利用湖南省重点实验室开放研究基金(MN2018K02)
收稿日期:2019-04-29;修订日期:2019-10-28
通信作者:李昌新,讲师,博士;电话:025-58139551;E-mail:lichangxin2010@njtech.edu.cn;lichangxin20160706@163.com

