���±�ţ�1004-0609(2011)02-0405-06
AuPd/C��Ϊֱ��NaBH4-H2O2ȼ�ϵ������
����������
�� �꣬�����ѣ������������������� ��, �����ã�����
(��̶��ѧ ��ѧѧԺ �����Ѻû�ѧ��Ӧ�ý������ص�ʵ���ң���̶ 411105)
ժ Ҫ�����ý��ջ�ԭ���Ʊ���ͬ������AuPd/C�������ӣ��ֱ����X��������(XRD)����羵(TEM)�Դ������нṹ����ò����������CHI660a�绯ѧ����վ�Դ������е绯ѧ���ԣ�����������������Ͼ�Ϊ���������ṹ��AuPd/C�����Ͻ����ӵ�����Ϊ5 nm���ң���Au/C�е�����Au���Ӹ�С���Ҿ��ȷ�ɢ��VXC-72R̿�ڵı��棻Au/C�ķ�����ܶ�Ϊ25.05 mA/cm2����Au/C��ȣ�AuPd/C�������NaBH4�ĵ����������ԣ�������AuPd/CΪ����������Au/CΪ���������Ƴ�ֱ��NaBH4-H2O2ȼ�ϵ��(DBHFC)��������Au1Pd2/CΪ����������DBHFCӵ�����õĵ�����ܣ����¶�Ϊ60 �桢NaBH4Ũ��Ϊ1 mol/LʱDBHFC��������ܶȴﵽ114.6 mW/cm2��
�ؼ��ʣ�ֱ��NaBH4-H2O2ȼ�ϵ�أ�AuPd/C���������ӣ���������
��ͼ����ţ�O643���� ���ױ�־�룺A
Performance of AuPd/C as anode catalyst of
direct NaBH4-H2O2 fuel cell
WANG Hong, WANG Xian-you, HE Pei-ying, YI Lan-hua, PEI Fu, LONG Wang-mei, LI Jiao-jiao
(Key Laboratory of Environmentally Friendly Chemistry and Applications, Ministry of Education,
School of Chemistry, Xiangtan University, Xiangtan 411105, China)
Abstract: The nanosized AuPd alloys were prepared by impregnation method. The structure and surface morphology were characterized by X-ray diffractometry (XRD) and transmission electron microscopy (TEM). The electrochemical performance was tested by a CHI660a Electrochemistry Workstation. The results show that the nanosized electrocatalyst particles have face cubic structure and uniformly distribute on the surfaces of the VXC-72R carbon black. The size of the AuPd alloys particles is about 5 nm, which is apparently less than that of the Au particles. The peak current density of Au/C is 25.05 mA/cm2 and compared to pure Au/C catalyst, AuPd/C catalysts distinctly enhance the catalytic activity for the direct oxidation of NaBH4. A simple direct borohydride-hydrogen peroxide fuel cell (DBHFC) is fabricated in which the Au/C is used as the cathode catalyst and the AuPb/C is used as the anode catalyst. The Au1Pd2/C catalytic anode presents the highest catalytic activity among the AuPd/C alloys studied and the DBHFC shows as high as 114.6 mW/cm2 power density at 1 mol/L NaBH4 and 60 ��.
Key words: direct borohydride-hydrogen peroxide fuel cell; AuPd/C; nanosized particles; anode electrocatalyst
ȼ�ϵ����һ�ָ�Ч��������Դ[1-3]��Ŀǰ��չ�Ƚ�Ѹ�ٵ�ֱ�Ӽ״�ȼ�ϵ��(DMFC)����Ч�ʵ͡������ܶȵͺͼ״���������[4-6]�������⻯��Ϊȼ�ϵ�ֱ�����⻯��ȼ�ϵ��(DBFC)�����ھ��б������ߡ������������Ⱦ��ȼ�����ڴ����������ŵ㣬����Ϊ��һ�ֺ��з�չDZ����ȼ�ϵ��[7-9]��
DBFC�ڼ��Խ��������¹������������缫�������£�
BH4- + 8OH- �� BO2- + 6H2O + 8e
 = -1.24 V (1)
= -1.24 V (1)
��ӦΪ�˵��ӹ��̣�Ȼ��NaBH4�ھ���������������Ϻ���ʵ�ְ˵���������CHATENET��[10-11]����BH4-�ڸ�����Au�ϱ��ֳ��ϸߵĵ��������ʣ��ӽ�������ֵ8������Au����BH4-��������Ӧ�Ĵ�����ȴ������[12]��GENG��[13]���ֶ�Ԫ������Ͻ�����ȵ��ʽ����������ֳ����ߵĴ����ԡ�Pd����BH4-�Ĵ����Ը߶�����������ȴƫ��[14]��Ϊ�˽����һ���⣬���Խ����ߵ��ŵ��ϣ��Ʊ�һ�ּȾ��иߴ������־��и߿���Ч�ʵĶ�Ԫ�������������о�������AuPd/C����Ч����H2���������ʣ�ʹ����Ч�ʴﵽ50%~70%[15]��
���������ý��ջ�ԭ��������ģ�巨�ֱ��Ʊ���̼��ʵ��Au��������(SAu/C)[16]��̼�ؿ���Au��������(HAu/C)[17]��������SAu/CΪ����������HAu/CΪ����������ɵ�ֱ��NaBH4-H2O2ȼ�ϵ��(DBHFC)��20 ���µ�������ܶ�Ϊ25.8 mW/cm2��
�ڴˣ��������߲��ý��ջ�ԭ���Ʊ���ͬĦ���ȵ�AuPd/C��������������������DBHFC�������������Դ����ĵ绯ѧ���ܼ�DBHFC�ĵ�����ܽ������о���
1 ʵ��
1.1 AuPd/C�������Ʊ�
��0.1 mol/L AuCl3��0.1 mol/L PdCl2��0.28 mL 0.1 mol/L PVP���μ���100 mL�ߴ�ˮ�У�����������Ũ��Ϊ0.001 mol/L������15 min�����������VXC-72R̿��(������̿�ڵ�������Ϊ20?80)����������30 min����3 mol/L��NaOH����pH��10���ң���μ���1 mL 1 mol/L NaBH4������2 h�����ˣ�������ʽ����������Ʊ�������ǰ��һ�¡�
1.2 AuPd/C������������ѧ���ܱ���
���ձ���ѧD/max-3C��X���������ǣ�����Ʒ����XRD���ԡ�Cu K������=0.154 056 nm��ʯī��ɫ��������100 mA����ѹ50 kV����FEI Tecnai G2(���ٵ�ѹΪ 200 kV)��羵(TEM) �۲���Ʒ����ò���۽ṹ��
1.3 �缫�Ʊ����绯ѧ���ܲ���
��ֱ��Ϊ3 mm�IJ�̿�缫����7#����ɰֽ�⣬����0.3 ��m��Al2O3������Ƥ���������棬�����ϴȥ�������Ȼ�����볬����ˮԡ����ϴ2~3 min������������Ҵ�(�����Ϊ1?1)��HNO3(�����Ϊ1?1)������ˮ��������ϴ��
��ȡ10 mg AuPd/C��Ʒ������0.25 mL 5% Nafion��Һ��0.75 mLȥ����ˮ����������ɢ2 h��Ȼ���������10 min������Һ��ȡ5 ��L����Һ���ڲ�̿�缫�ϣ����º͵ĵ������и��
�绯ѧ���ܲ���ʵ����þ�������缫��ϵ����AuPd/CΪ�����缫����ĭ��Ϊ�Ե缫��Ag/AgCl (3 mol/L KCl)Ϊ�αȵ缫ͨ���������ӣ����ҺΪ3 mol/L NaOH+0.1 mol/L NaBH4�����Һ��ɨ���ٶ�20 mV/s����������ΪCHI660a�绯ѧ����վ(�Ϻ�����������˾)��
1.4 ����Ʊ�
���Ʊ���AuPd/C��5% Nafion��Һ��������93:7 ���������������Ͼ��ȳɺ�״, ����״���ʾ��ȵ�Ϳ�ڸ�����, AuPd/C��Ϳ����Ϊ4 mg/cm2�� ��Ƭ���������16 MPa��ѹ1 min��
��AuPd/CΪ����������Au/CΪ����������Nafion117ĤΪ�����Ĥ����ͼ1��װ��ģ���ء��������ҺΪ2 mol/L H2O2+0.5 mol/L H2SO4�����Һ���������ҺΪ3 mol/L NaOH+1 mol/L NaBH4�����Һ�����Һ�����䶯��ѭ�����¡���صĸ��ص�����0 �仯��160 mA, ÿ��������������2 min���ڲ��Թ����У�������һ����������ϵر仯����һ��������
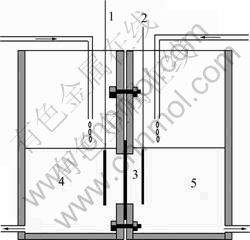
ͼ1 ֱ��NaBH4-H2O2ȼ�ϵ��ģ��ʾ��ͼ
Fig.1 Schematic diagram of direct NaBH4-H2O2 fuel cell: 1��Anode catalyst; 2��Cathode catalyst; 3��Activated Nafion 117 membrane; 4��1 mol/L NaBH4 in 3 mol/L NaOH; 5��2 mol/L H2O2 in 0.5 mol/L H2SO4
2 ���������
2.1 AuPd/C�����Ľṹ����ò����
ͼ2(a)��ʾΪAu/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C��XRD�ס�ͼ��2��=25��λ�ø����Ŀ������ΪVulcan XC-72R̿��(002)�������������塣2��Ϊ38.23�㡢44.33�㡢64.59�㡢77.51�㡢81.66��������ֱ��Ӧ��Au(JCPDF)��Ƭ��Au(111)��Au(200)��Au(220)��Au(300)��Au(222)�������塣3��AuPd������XRD����Au�����ơ�2��Ϊ39.18�㡢45.73�㡢67.34�㡢81.16�㡢86.09��������ֱ��Ӧ��Pd(JCPDF)��Ƭ��Pd(111)��Pd(200)��Pd(220)��Pd(300)��Pd(222)�������塣˵�����ý��ջ�ԭ���Ʊ��Ĵ�����Ϊ������������ͼ2(b)��ʾΪ�Ը�������Au��Pd��(111)�����֮���XRD�Ľ�����3��AuPd/C��������ʾ��˫�壬�������Au(111)��Pd(111)�����������λ�ƣ���ʾ���мȺ��е���Au��Ҳ����AuPd�Ͻ�������ΪAu�Ļ�ԭ����Ҫ����Pd���ڼ��뻹ԭ��NaBH4��Au���ȱ���ԭ��
ͼ3��ʾΪAu/C(a)��Au1Pd2/C(b)��TEM��ͼ3(a)���Կ�����ֱ��10 nm���ҵ�Au���Ӿ��ȵķ�ɢ��Vulcan XC-72R̿�ڱ��棬��Au1Pd2�Ͻ����ӵ�ֱ��ֻ��5 nm���ң�С��Au���ӵģ��ҷ�ɢ���ȡ�
2.2 �绯ѧ���ܷ���
2.2.1 ����ɨ���������
ͼ4��ʾΪAu/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C��3 mol/L NaOH��0.1 mol/L NaBH4�����Һ�е�����ɨ��������ߣ�ɨ��Ϊ20 mV/s����ͼ4�п��Կ�����Au/C��-0.6~0.4 V (vs Ag/AgCl)֮����ֿ������壬��GYENGE[18]��������BH4-��Au�ı�����а˵��ӷ�Ӧ(Eq.(1))�ķ���ͼһ�£�˵��BH4-��Au/C�����ϵ�������ӦΪֱ�Ӱ˵���������Au/C�ķ�����ܶ�Ϊ25.05 mA/cm2����Pd/C�ķ�����ܶ�Ϊ34.29 mA/cm2����Au/C�����36.9 %����ʾ����Au/C���ߵĴ����ԡ���3�ֲ�ͬ������AuPd/C�����ķ�����ܶ���Pd/C���ƣ�˵����ʹֻ�Ǽ���������PdҲ����Ч��ߴ����Ĵ����ԡ�ͬʱ����ͼ�л����Կ�����AuPd/C�����ķ��λҲ��Au/C��Pd/C�ĸ���������AuPd/C�����ܽ���BH4-����������λ��������������Ӧ�Ľ��С�
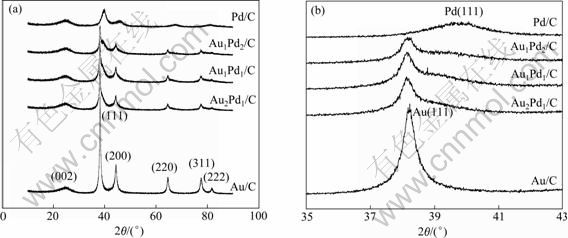
ͼ2 Au/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C��XRD���ֲ��Ŵ�ͼ
Fig.2 XRD patterns of Au/C, Au2Pd1/C, Au1Pd1/C, Au1Pd2/C and Pd/C(a) and their partically enlarged views(b)
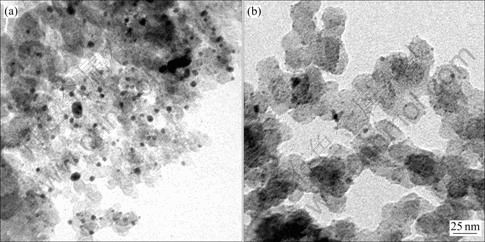
ͼ3 Au/C��Au1Pd2/C��TEM��
Fig.3 TEM images of Au/C (a) and Au1Pd2/C (b)
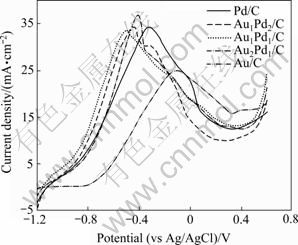
ͼ4 Au/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C������ɨ�����ͼ
Fig.4 Linear scanning voltammograms of Au/C, Au2Pd1/C, Au1Pd1/C, Au1Pd2/C and Pd/C electrodes
2.2.2 ��ʱ��λ����
��ʱ��λ�����ǶԵ�������Է�������Ч���ߣ��ܹ�ģ���ع����ĺ������ͼ5��ʾAu/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C��3 mol/L NaOH��0.1 mol/L NaBH4�У��㶨����Ϊ8.5 mA/cm2�ļ�ʱ��λͼ����ͼ5�ɿ�����120 s��Au/C�Ĺ�����λΪ-0.724 V������Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C�缫�ϵĹ�����λ�ֱ��Au/C�缫�ĸ�0.138��0.116��0.176��0.08 V��˵����AuPd/C������BH4-����������λҪ����Au/C��Pd/C�ġ���3��AuPd/C�����У�Au1Pd2/C��BH4-�ĵ�������������ߡ�
2.3 ������ܷ���
2.3.1 ����Au��Pd������Ӱ��
ͼ6��ʾΪ�ֱ���Au/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C��Ϊ������������Au/C��Ϊ�����������DBHFC�ĵ�ؼ������ߺ����ܶȣ���ع����¶�Ϊ20 �档��ͼ6(a)�пɼ������ŵ����ܶȵ�����DBHFC�ĵ�ص�ѹ�����½�����0~10 mA/cm2��Χ�ڣ���Au/C��Ϊ����������DBHFC�ĵ�ص�ѹ�½����ʱȽ����ԡ���Ϊ�ڴ˹����У���������ܵ绯ѧ������Ӱ�������Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C��Ϊ����������DBHFC�ĵ�ص�ѹ���ŵ����ܶȵ�������������½����ƣ����������Ҫ��ŷķ������Ӱ�졣����Pd����������صĿ�·��ѹ����������Au2Pd1/C��Au1Pd1/C��Au1Pd2/CΪ����������DBHFC��������ܶȾ�����Au/C��Pd/C�ġ�Au1Pd2/CΪ����������DBHFCӵ�нϺõĵ�����ܣ�������ܶȼ���Ӧ�ĵ����ܶȷֱ�Ϊ56.8 mW/cm2��70 mA/cm2��
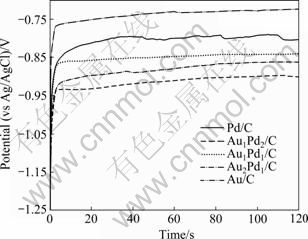
ͼ5 Au/C��Au2Pd1/C��Au1Pd1/C��Au1Pd2/C��Pd/C�ļ�ʱ��λ����
Fig.5 Chronopotentiometry curves of Au/C, Au2Pd1/C, Au1Pd1/C, Au1Pd2/C and Pd/C

ͼ6 ��ͬ����������DBHFC��ؼ������ߺ����ܶ�����
Fig.6 Cell polarization curves (a) and power density curves (b) of DBHFC using various anode catalysts
2.3.2 NaBH4Ũ�ȵ�Ӱ��
ͼ7��ʾΪ��Au1Pd2/CΪ����������DBHFC��NaBH4Ũ�ȶ������ܵ�Ӱ�졣��ͼ7�ɼ����������ܶȴ���35 mA/cm2ʱ��0.1 mol/L NaBH4��DBHFC���ܼ����������Ϊ�ڵ�Ũ��ʱ�������ܶ�����һ���̶ȣ��������������������ݣ��谭��BH4-��缫����ɢ��ʹ�õ缫����������������NaBH4Ũ�ȵ�������һ����õ����⣬��������ܶ�Ҳ��֮������NaBH4Ũ�ȼ�������ʱ��������ܷ������͡�����Ũ�ȵ�NaBH4ʹ����Au1Pd2/C�����BH4-����Զ�����������������ĵ����������´���BH4-û�б������Dz���ˮ�ⷴӦ����NaBH4Ũ�ȵ�����Ҳʹ���������Һ�������ӣ�Ӱ��BH4-����ɢ���ʣ����ӵ�ص�ŷķ��ģ����͵�����ܡ���ˣ�NaBH4Ũ��Ϊ1 mol/L ��DBHFC���ܽϺá�
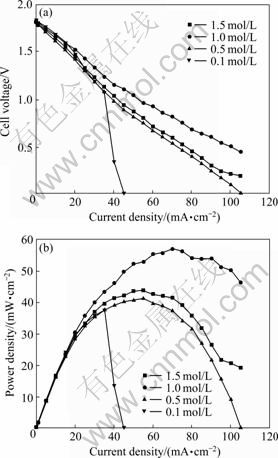
ͼ7 NaBH4Ũ�ȶ�DBHFC���ܵ�Ӱ��
Fig.7 Relationship of DBHFC cell performance with concentrations of NaBH4: (a) Curves of cell voltage vs current density; (b) Curves of power density vs current density
2.3.3 �¶ȶ�DBHFC���ܵ�Ӱ��
һ����Ϊ��DBHFC�����������¶ȵ����߶����(��ͼ8)���¶ȵ�������������ӵ��˶��ٶ�,Ҳ�����BH4-����������,��صĵ�ѹ�������¶����߶�����,�ϸ��¶�ʱ��ر��ֳ��Ϻõĵ�����ܡ�������Ҳ�����BH4-��ˮ�����ʣ�Ҳ��������BH4-�Ĵ���ǰ�ή��ȼ�ϵ������ʣ�����Ӱ���������ϵĴ����ԡ���ʵ��������¶�Ϊ20��30��40��50��60 ���DBHFC�ĵ�����ܡ������60 ��ʱ������ܶ�Ϊ114.6 mW/cm2����Ӧ�ķ�����ܶ�Ϊ107.5 mA/cm2��
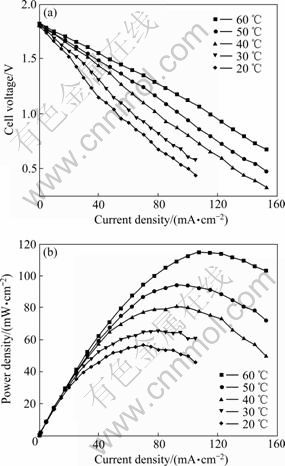
ͼ8 �¶ȶ�DBHFC���ܵ�Ӱ��
Fig.8 Effect of temperature on cell performance of DBHFC: (a) Curves of cell voltage vs current density; (b) Curves of power density vs current density
3 ����
1) ���ý��ջ�ԭ���Ʊ�������Ϊ5 nm���ҵ�����������������AuPd/C, ��ֱ��С��Au/C�е�����Au���ӣ��Ҿ��ȷ�ɢ��VXC-72R̿�ڵı��档
2) ������ɨ����������У�Au/C�ķ�����ܶ�Ϊ25.05 mA/cm2��ֻ������Pd��AuPd/C�����ķ�����ܶ���Pd/C�����ƣ���Au/C�����36.9%����ʾ�����ߵĵ�����ԡ���AuPd/C�����ķ��λҲ��Au/C��Pd/C�ĸ���������AuPd/C�����ܽ���BH4-����������λ��������������Ӧ�Ľ��С����ҷ�����3��AuPd/C�����У�Au1Pd2/C��BH4-�ĵ�������������ߡ�
3) ��Au2Pd1/C��Au1Pd1/C��Au1Pd2/CΪ����������DBHFC��������ܶȾ�����Au/C��Pd/C�ġ�Au1Pd2/CΪ����������DBHFCӵ�нϺõĵ�����ܣ�������ܶȼ���Ӧ�ĵ����ܶȷֱ�Ϊ56.8 mW/cm2��70 mA/cm2��
4) NaBH4Ũ�ȹ�����;���Ӱ��DBHFC�ĵ�����ܣ� NaBH4Ũ��Ϊ1 mol/LʱDBHFC�ĵ�����ܽϺá��������������DBHFC�ĵ�����ܣ������60 ��ʱ��������ܶ�Ϊ114.6 mW/cm2����Ӧ�ķ�����ܶ�Ϊ107.5 mA/cm2��
REFERENCES
[1] APPLEBY A J. Fuel cell technology: Status and future prospects[J]. Energy, 1996, 21(7/8): 521-653.
[2] �ſ���, �� ��, �� �D, ��ΰ��. ���ӽ���Ĥȼ�ϵ�ص�������о���״��չ��[J]. �����, 2010, 31(2): 67-73.
ZHANG Jun-min, WEN Ming, LI Yang, GUAN Wei-ming. Research status and prospect of electro-catalyst for proton exchange membrane fuel cell[J]. Precious Metals, 2010, 31(2): 67-73.
[3] WEE J H. A comparison of sodium borohydride as a fuel for proton exchange membrane fuel cells and for direct borohydride fuel cells[J]. Journal of Power Sources, 2006, 155(2): 329-339.
[4] SCHULTZ T, ZHOU S, SUNDMACHER K, SCOTT K, GINKEL M, GILLES E D. Dynamics of the direct methanol fuel cell (DMFC): Experiments and model-based analysis[J]. Chemical Engineering Science, 2001, 56(2): 333-341.
[5] APANEL G, JOHNSON E. Direct methanol fuel cells-ready to go commercial? [J]. Fuel Cells Bulletin, 2004(11): 12-17.
[6] ��־��, ��Ҫ��, л����, ����ΰ, ������. PtRuP/�������Ʊ������[J]. ���ϴ�ѧѧ��: ��Ȼ��ѧ��, 2008, 39(3): 448-453.
YANG Zhi-kuan, WANG Yao-wu, XIE Xiao-feng, GUO Jian-wei, HU Guo-rong. Preparation and characterization of PtRuP/C catalysts[J]. Journal of Central South University: Science and Technology, 2008, 39(3): 448-453.
[7] MA Jia, CHOUDHURY N A, SAHAI Y. A comprehensive review of direct borohydride fuel cells[J]. Renewable and Sustainable Energy Reviews, 2010, 14(1): 183-199.
[8] AMENDOLA S C, ONNERUD P , KELLY M T, PETILLO P J, SHARP-GOLDMAN S L, BINDER M. A novel high power density borohydride-air cell[J]. Journal of Power Sources, 1999, 84(1): 130-133.
[9] κ����, ������, ������, ������, �� ��. ֱ�����⻯��ȼ�ϵ��[J]. ��ѧ��չ, 2008, 20(9): 1427-1432.
WEI Jian-liang, WANG Xian-you, YI Si-yong, DAI Chun-ling, LI Na. Direct borohydride fuel cell[J]. Progress in Chemistry, 2008, 20(9): 1427-1432.
[10] CHATENET M, MICOUD F , ROCHE I , CHAINET E, J VONDRAK. Kinetics of sodium borohydride direct oxidation and oxygen reduction in sodium hydroxide electrolyte: Part ��. BH4- electro-oxidation on Au and Ag catalysts[J]. Electrochim Acta, 2006, 51(25): 5459-5467.
[11] CHENG H, SCOTT K. Determination of kinetic parameters for borohydride oxidation on a rotating Au disk electrode[J]. Electrochimica Acta, 2006, 51(17): 3429-3433.
[12] MASEL R I. Chemical kinetics and catalysis[M]. New York: Wiley-Interscience, 2001: 667/755, 837-880.
[13] GENG Xiao-ying, ZHANG Hua-min, YE Wei, MA Yuan-wei, ZHONG He-xiang. Ni-Pt/C as anode electrocatalyst for a direct borohydride fuel cell[J]. Journal of Power Sources, 2008, 185(2): 627-632.
[14] LIU Bin-hong, LI Zhou-peng, SUDA S. Electrocatalysts for the anodic oxidation of borohydrides[J]. Electrochimica Acta, 2004, 49(19): 3097-3105.
[15] PARK S Y, LEE D W, PARK I S, HONG Y K, PARK Y M, LEE K Y. The effective bimetallic component of Pd-Au/C for electrochemical oxidation of borohydrides[J]. Current Applied Physics, 2010, 10(2): S40-S43.
[16] κ����, ������, �� ��, ��˳��, ������, �� ��. ����Au������Ϊֱ�����⻯��-��������ȼ�ϵ����������[J]. ��ѧѧ��, 2008, 66(24): 2675-2680.
WEI Jian-liang, WANG Xian-you, WANG Hong, YANG Shun-yi, DAI Chun-ling, PEI Fu. Performance of nanosized Au particle as a cathode catalyst for direct borohydride-hydrogen peroxide fuel cell[J]. Acta Chimica Sinica, 2008, 66(24): 2675-2680.
[17] WEI Jian-liang, WANG Xian-you, WANG Ying, CHEN Quan-qi, PEI Fu, WANG Yan-sheng. Investigation of carbon-supported Au hollow nanospheres as electrocatalyst for electrooxidation of sodium borohydride[J]. International Journal of Hydrogen Energy, 2009, 34(8): 3360-3366.
[18] GYENGE E. Electrooxidation of borohydride on platinum and gold electrodes: implications for direct borohydride fuel cells[J]. Electrochimica Acta, 2004, 49(6): 965-978.
(�༭ ������)
������Ŀ������ʡ�Ƽ����ص���Ŀ(2009WK2007)������ʡ��Ȼ��ѧ�������ϻ����ص���Ŀ(09JJ8001)
�ո����ڣ�2010-02-22�������ڣ�2010-11-16
ͨ�����ߣ������ѣ����ڣ���ʿ���绰��0731-58292060��E-mail: wxianyou@yahoo.com

