DOI��10.19476/j.ysxb.1004.0609.2019.02.19
��(�������)ԭ����λ���Ӷ�˫���-TiAl/��2-Ti3Al��������������Ժ͵������ʵ�Ӱ��
���칦1, 2��������1���絤��2��������1����ѩ��2
(1. �й���ѧ ��ѧԺ��ά�����뼼���о�������� 300300��
2. �й���ѧ ��ŷ���չ���ʦѧԺ����� 300300)
ժ Ҫ�����û����ܶȷ������ۼ����о�����(�������)ԭ����λ����˫���-TiAl/��2-Ti3Al������ϵ��ƽ���γ��ܡ����ѹ������ӽṹ�ȡ������ʾ������������ϵ����������ƽ���γ��ܾ�Ϊ��ֵ���������ǿ�����ʵ���Ʊ������ȶ����ڡ��Դ�������ϵ�Ķ��ѹ���̬�ܶȷ�����������ϵMo-Sa5 (��Cr-Sa5)�Ľ��ǿ�ȼ�����ͬʱ��ϵMo-Sa5��Mo-d��Ti-d����̬�ܶ����ӡ��������ǿ����Ti-d��Al-p����ӻ���ǿ�Ƚ��ͣ�λ���˶����������٣������ڸ���TiAl�Ͻ���ϵ����ԡ�������ԭ������(001)�����ܶȺͲ������ķ������֣�Mo(��V)�IJ�������������������ԭ����Χ�ľۼ�ЧӦ���γɽ��ǿ���Ըߵ�������������Χ���������ϵĸ������Գ̶��½��������Ǵ���TiAl�Ͻ����Ը��Ƶ�����
�ؼ��ʣ�˫���-TiAl/��2-Ti3Al���棻�������ʣ����ԣ��������ʣ���һ��ԭ��
���±�ţ�1004-0609(2019)-02-0370-10���� ��ͼ����ţ�TG146.2���� ���ױ�־�룺A
TiAl�Ͻ���������۵㡢���ܶȡ��ߵ���ģ�������õĿ������Ϳ�������ܣ��������ں��պ������������������ľ�DZ��[1-2]��Ȼ������������չ�Ժ����Խϲ�ӹ����������������������Ĺ㷺Ӧ��[3-5]����ˣ�Ŀǰ�о����ص����ڱ������������ܵ�ǰ���£����TiAl�Ͻ��������չ�Ժ����ԡ���Ϊ��-TiAl���Ͻ�ڶ��࣬��2-Ti3Al�������ܹ�������Ͻ������[6-8]��˫��TiAl-Ti3Al�Ͻ�������������ǿ�ȺͿ��������ܱ��ܹ�ע[9-10]�����ʹ��������о��������롢ʵ��̽������ϸ�£����ɶ��ڽ�ʾTiAl/Ti3Al����ı��λ��������TiAl�Ͻ�����Ծ�����Ҫ���壬ҲΪ�з����ܸ�Ϊ������²����ṩ�˻������ݡ�
��������Ϊ�����TiAl�Ͻ���ۺ����ܣ��о���Ա�����˴���������̽����ʵ���о������ֺϽ��Ͻ��ǽ�����Ͻ���ԣ�������Ժ����Ե���Ҫ;��[11-14]��ʵ���о����������ڦ�-TiA����ԣ���(111)ƽ��������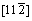 ����Ļ����������ģ�����a2-Ti3Al�࣬��(0001)ƽ��������
����Ļ����������ģ�����a2-Ti3Al�࣬��(0001)ƽ��������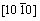 ����Ļ�����������[15-16]��ףӨӨ��[17]��С�ͷ��Ժ��ټ��绡¯�����Ʊ���Ti50Al48Cr2�Ͻ�����������û�����о�����Cr����ʹ�Ͻ���壬�ر��Ǿ��紦�ĵ���̬�ܶ����ӡ���������Cr�ļ���ʹ�Ͻ���d-d�����������ǿ���Ӷ������˺Ͻ�����ԡ������[18]ͨ��ʵ�����������CrԪ�ض�TiAl�Ͻ��������Ժ��۽ṹ��Ӱ�죬����CrԪ�ص����ӽ�����TiAl�Ͻ�ĶѶ����ܣ�ʹ�úϽ���λ����������ʱ��������������ʽ�������Ա��Σ��Ӷ���ýϸߵ����ԣ�������3% Cr��TiAl�Ͻ������������ѣ�ͬʱ���ֺϽ�ľ������Լ�Ti3Al�ڶ���ĺ�������������Ҳ��Ӱ�졣�����ϣ��������[19]�о����ڦ�-TiAl�Ͻ��м���MoԪ�أ�������Ũ��Ϊ12.5%ʱ��Mo�����ܽϺõظ���TiAl�Ͻ�����ԣ�������Tiԭ�ӵ�s��p��d�������ڽ���Moԭ�ӷ�����ǿ�ҵ�s-s��p-p��d-d��������ã���Ч�������˺Ͻ���Ti��Alԭ�ӵ�Ǩ�ƣ���������ߺϽ���ȶ��Ժ�ǿ�ȡ�κǿ��[20]����Ƕ��ԭ�ӷ��������о������ֵ�������Mo��WԪ�ػ����Ti3Al�Ͻ��Ӳ�ȣ�Cr���Tiλ������ӺϽ��Ӳ�ȡ���������[21]�о���Nb��Mo�ϽԦ�-TiAl�Ͻ��Ӱ�죬�������Nb��Mo�����������Ԫ��������ڻ����Ԫ��֮���������Լ���Ӧԭ��֮��ļ���ǿ�ȣ����½�ǿ�Ĺ���ǿ��ЧӦ���Ӷ�Ӱ���-TiAl����ѧ���ʡ�MORINAGA��[22]�о�������TiAl�����д���Al-p(Alԭ�ӵ�3p���)��Ti-d(Tiԭ�ӵ�3d���)����ӻ��ķ����Լ���, ʹ���ϵļ���ǿ����ߣ�����������������Խϲ����ͨ�����ӵ���Ԫ������ǿTi��Ti��������Al��Ti�������Ը��ƺϽ�����ԡ�HAO��[23]���õ�һ��ԭ�����������о��˦�-TiAl�ͦ�2-Ti3Al��V��Cr��Mn��Fe��Ni��Zr��Nb��Mo��Ta��Ga��Sn��λ��ռ������������ʾ���ڦ�-TiAl���У�Zr��Nb��Taԭ������ռ��Tiλ����Fe��Ni��Ga��Snԭ������ռ��Alλ����һ���棬V��Cr��Mnԭ��λ�õ�ѡ��ȡ���ںϽ����ɡ��ڦ�2-Ti3Al���У�Ga��Snԭ������ѡ��Alλ����V��Cr��Mn��Zr��Nb��Mo��Taԭ������ռ��Tiλ������������������ڶ��ڵ���TiAl���Ͻ�ṹ�����Ե����ʵ�̽�����ԺϽ�Ԫ�ص�ռλ���Ҳ���ڲ�һ�µĽ��ۡ����ߣ�TiAl�Ͻ���ڶ�����ṹ���Ƚϵ��͵��Ǧ�-TiAl��ͦ�2-Ti3Al��棬�����о����֣�������2��Ĵ����ܸ��Ʋ��ϵ����Ի�����[24-26]��WEI��[24]���õ�һ��ԭ��������Ͼ����оݣ��о��˹��ɽ���V��Cr��Nb��Ta��W��Re��˫��TiAl-Ti3Al�Ͻ�Ľ������ʵ�Ӱ�죬���������˽���Ľ����ܣ������˲��ȶ�����ܣ���������ڸ���TiAl/Ti3Al��������ԡ�������[25]�ӵ��ӽṹ��ν���̽������Ϊ����洦�ļ����ǿ���������������ӽ����ȶ����������ԡ���ߺϽ�����ԡ���ΰ����[26]������Mn���ӵ�˫��TiAl�Ͻ���棬���ڲ�Ƭ״�ṹ�������Ե��ۻ��ơ�����δ�������ںϽ�Ԫ�ضԴ���˫��Ͻ�Ľ�����ȶ��Ժ͵������ʵĶ������о��������������֪���Ͻ���ϵ�����ȡ�������۽ṹ�����۽ṹ��������ʽ�����ء�
����Ļ�����������[15-16]��ףӨӨ��[17]��С�ͷ��Ժ��ټ��绡¯�����Ʊ���Ti50Al48Cr2�Ͻ�����������û�����о�����Cr����ʹ�Ͻ���壬�ر��Ǿ��紦�ĵ���̬�ܶ����ӡ���������Cr�ļ���ʹ�Ͻ���d-d�����������ǿ���Ӷ������˺Ͻ�����ԡ������[18]ͨ��ʵ�����������CrԪ�ض�TiAl�Ͻ��������Ժ��۽ṹ��Ӱ�죬����CrԪ�ص����ӽ�����TiAl�Ͻ�ĶѶ����ܣ�ʹ�úϽ���λ����������ʱ��������������ʽ�������Ա��Σ��Ӷ���ýϸߵ����ԣ�������3% Cr��TiAl�Ͻ������������ѣ�ͬʱ���ֺϽ�ľ������Լ�Ti3Al�ڶ���ĺ�������������Ҳ��Ӱ�졣�����ϣ��������[19]�о����ڦ�-TiAl�Ͻ��м���MoԪ�أ�������Ũ��Ϊ12.5%ʱ��Mo�����ܽϺõظ���TiAl�Ͻ�����ԣ�������Tiԭ�ӵ�s��p��d�������ڽ���Moԭ�ӷ�����ǿ�ҵ�s-s��p-p��d-d��������ã���Ч�������˺Ͻ���Ti��Alԭ�ӵ�Ǩ�ƣ���������ߺϽ���ȶ��Ժ�ǿ�ȡ�κǿ��[20]����Ƕ��ԭ�ӷ��������о������ֵ�������Mo��WԪ�ػ����Ti3Al�Ͻ��Ӳ�ȣ�Cr���Tiλ������ӺϽ��Ӳ�ȡ���������[21]�о���Nb��Mo�ϽԦ�-TiAl�Ͻ��Ӱ�죬�������Nb��Mo�����������Ԫ��������ڻ����Ԫ��֮���������Լ���Ӧԭ��֮��ļ���ǿ�ȣ����½�ǿ�Ĺ���ǿ��ЧӦ���Ӷ�Ӱ���-TiAl����ѧ���ʡ�MORINAGA��[22]�о�������TiAl�����д���Al-p(Alԭ�ӵ�3p���)��Ti-d(Tiԭ�ӵ�3d���)����ӻ��ķ����Լ���, ʹ���ϵļ���ǿ����ߣ�����������������Խϲ����ͨ�����ӵ���Ԫ������ǿTi��Ti��������Al��Ti�������Ը��ƺϽ�����ԡ�HAO��[23]���õ�һ��ԭ�����������о��˦�-TiAl�ͦ�2-Ti3Al��V��Cr��Mn��Fe��Ni��Zr��Nb��Mo��Ta��Ga��Sn��λ��ռ������������ʾ���ڦ�-TiAl���У�Zr��Nb��Taԭ������ռ��Tiλ����Fe��Ni��Ga��Snԭ������ռ��Alλ����һ���棬V��Cr��Mnԭ��λ�õ�ѡ��ȡ���ںϽ����ɡ��ڦ�2-Ti3Al���У�Ga��Snԭ������ѡ��Alλ����V��Cr��Mn��Zr��Nb��Mo��Taԭ������ռ��Tiλ������������������ڶ��ڵ���TiAl���Ͻ�ṹ�����Ե����ʵ�̽�����ԺϽ�Ԫ�ص�ռλ���Ҳ���ڲ�һ�µĽ��ۡ����ߣ�TiAl�Ͻ���ڶ�����ṹ���Ƚϵ��͵��Ǧ�-TiAl��ͦ�2-Ti3Al��棬�����о����֣�������2��Ĵ����ܸ��Ʋ��ϵ����Ի�����[24-26]��WEI��[24]���õ�һ��ԭ��������Ͼ����оݣ��о��˹��ɽ���V��Cr��Nb��Ta��W��Re��˫��TiAl-Ti3Al�Ͻ�Ľ������ʵ�Ӱ�죬���������˽���Ľ����ܣ������˲��ȶ�����ܣ���������ڸ���TiAl/Ti3Al��������ԡ�������[25]�ӵ��ӽṹ��ν���̽������Ϊ����洦�ļ����ǿ���������������ӽ����ȶ����������ԡ���ߺϽ�����ԡ���ΰ����[26]������Mn���ӵ�˫��TiAl�Ͻ���棬���ڲ�Ƭ״�ṹ�������Ե��ۻ��ơ�����δ�������ںϽ�Ԫ�ضԴ���˫��Ͻ�Ľ�����ȶ��Ժ͵������ʵĶ������о��������������֪���Ͻ���ϵ�����ȡ�������۽ṹ�����۽ṹ��������ʽ�����ء�
������Mo��Cr��V�ֱ����˫���-TiAl/��2-Ti3Al������ϵ��Ϊ�о��������ܶȷ������ۿ�����о��Ͻ���ϵ�ľ���ṹ���ȶ��Ժ����ԣ�̽������Ͻ��������������ʵ����ڹ�ϵ��ּ�ڽ�ʾ˫��TiAl�Ͻ����Ի����Ը��Ƶ��ۻ��ơ�Ԥ���������ܣ�Ϊ����Ͻ���ϵ�ʵ���о��ṩ�������ݡ�
1 ����ģ������㷽��
1.1 ����ģ�ͼ���ѡ��
Ϊ���о�˫��TiAl���Ͻ���ϵ�����Ƚ�����-TiAl�ͦ�2-Ti3Al�Ľṹģ�͡���-TiAlΪL10�������ķ��ṹ���ռ�ȺΪP4/mmm���侧���а���2��Tiԭ�Ӻ�2��Alԭ�ӣ��������Ϊa=b=0.400 nm��c=0.406 nm����ͼ1(a)��ʾ����2-Ti3AlΪD019�;���ṹ���������Ϊa =b=0.576 nm��c=0.466 nm����ͼ1(b)��ʾ��
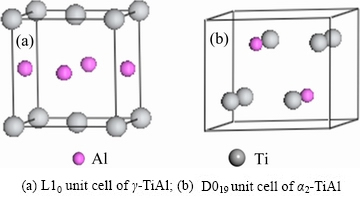
����˫��TiAl/Ti3Al������ϵ���ԣ��佨�������Ϊ���ӡ�����ÿ�־���ı�����ֶ�������-TiAl�ͦ�2-Ti3Al�ı���������ϣ��ʹ��ڶ��ֿ��ܵĽ���ģ�͡������������ȶ��Ժã�����ѡ���-TiAl 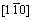 (111)����2-Ti3Al
(111)����2-Ti3Al 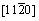 (0001)��������������ģ�͡���TiAl�Ͻ��е�ȡ���ϵ�Ѿ�������������۲�ʵ����֤ʵ[16]��Ϊ�˾������ͽ���ģ�͵Ĵ���ȣ��Ԧ�-TiAl (111) 4�����ṹ��Ԫ�ͦ�2-Ti3Al (0001) 5�����ṹ��ԪΪ����������1��2��1�ij����ṹ���������ϲ����10
(0001)��������������ģ�͡���TiAl�Ͻ��е�ȡ���ϵ�Ѿ�������������۲�ʵ����֤ʵ[16]��Ϊ�˾������ͽ���ģ�͵Ĵ���ȣ��Ԧ�-TiAl (111) 4�����ṹ��Ԫ�ͦ�2-Ti3Al (0001) 5�����ṹ��ԪΪ����������1��2��1�ij����ṹ���������ϲ����10 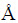 ����ղ㡣�������ϵͳ����Tiԭ��46����Alԭ��26������-TiAl (111)�ͦ�2-Ti3Al (0001)���γ������ʱ��������ֲ�ͬ�Ľṹ�����Ӧ��ģ����ͼ2��ʾ��
����ղ㡣�������ϵͳ����Tiԭ��46����Alԭ��26������-TiAl (111)�ͦ�2-Ti3Al (0001)���γ������ʱ��������ֲ�ͬ�Ľṹ�����Ӧ��ģ����ͼ2��ʾ��
ͼ1 ��-TiAl�ṹģ�ͺͦ�2-Ti3Al�ṹģ��
Fig. 1 Structure models of TiAl
ͼ2 ��-TiAl/��2-Ti3Al����ģ��
Fig. 2 ��-TiAl/��2-Ti3Al interface models
Ϊ��ѡȡ���ʵ�ģ������㷽�����ȶԦ�-TiAl�ͦ�2-Ti3Alģ�͵�Ԫ�����Ż����õ��Ż���������뱨����ʵ����(���ڦ�-TiAl��a=b=0.400 nm��c0=0.408 nm�����ڦ�2-Ti3Al��a=b=0.577 nm��c0=0.462 nm)[27-29]���бȽϡ���������ʵ������һ������Χ���Ǻϵúܺã������ʹ�õ�ģ�ͺͼ��㷽�����С��Դ˼��㷽����ͼ2��ʾ������������ϵ���м����Ż����õ��������ֱ�Ϊ-75233.8186 eV��-75234.1603 eV���ɼ���ģ��2�Ľ���ܸ��ͣ��ṹ���ȶ��������ļ���ѡ�ô�ģ�͡�
Ϊ���о�Mo��Cr��VԪ�ض�S0(˫���-TiAl/��2- Ti3Al)�Ͻ���ϵ�Ľṹ�����ʵ�Ӱ�죬��Mo��Cr��V�ֱ����Ti(��Al)��������˫���-TiAl/��2-Ti3Al�Ͻ���ϵģ�͡��������Ti��λ����5�֣��ֱ�Ϊa1������a5�����Al��λ����3�֣��ֱ�Ϊb1��b2��b3�����ǵ�λ����ͼ2(b)��ʾ��������ÿ�ֺϽ�Ԫ�ز��ӣ�������8������˫���-TiAl/��2-Ti3Al�Ͻ���ϵ�������Tiԭ�ӵ���ϵSa1������Sa5�����Alԭ�ӵ���ϵSb1��Sb2��Sb3����24������˫���-TiAl/��2-Ti3Al������ϵ��
1.2 ���㷽��
���û����ܶȷ������۵ĵ�һ��ԭ��[30-31]������������뻯ѧ���۷��������о�������ƽ�沨���Ʒ�����ѡ������ݶȽ�����PBE�������������������ã��ó���������������ʵ��۵���֮���������ơ�ƽ�沨�ض�������Ϊ350 eV������Ԩ��k������Ϊ4��2��1������Pulay�ܶȻ�Ϸ�������Ǣ����(SCF)�������������������Ϊ��ԭ�ӵ������仯��Ϊ2.0��10-5 eV / atom��ԭ��������Ϊ0.05 eV / nm��ԭ��λ�����Ϊ0.002 ��Ӧ��ƫ��Ϊ0.1 GPa��
����ƽ�沨���Ʒ�����CASTEP�����������ø����ܼ������Ⱥ��ɼ������о�������ţ���㷨�Ը���˫��TiAl/Ti3Al������ϵ���м����Ż���ֱ���ﵽ��������������ƽ��״̬�¾���Ľṹ����������������ɷֲ������������ڴ˻����ϣ�������������ѧ���۽��з��������ۡ�
2 �����������
2.1 ���Ӷ�������ȶ��Ե�Ӱ��
Mo��Cr��VԪ�����������Tiԭ�ӻ�Alԭ�Ӻ�����ϵ�ľ�������������仯������ϵ���ȶ�����һ����Ӱ�졣Ϊ�˷�����������ı仯�Ԧ�-TiAl/��2-Ti3Al������ϵ�ȶ��Ե�Ӱ�죬�ڼ���������ͬ������£��Խ���Ľṹģ�ͽ������о�����1�г��˸���������ϵ�ṹ�Ż���Ľ����
��1 Mo��Cr��V���Ӧ�-TiAl/��2-Ti3Al������ϵ�ļ������ʺ���������
Table 1 Geometrical and energy properties of Mo, Cr and V-doping ��-TiAl/��2-Ti3Al interface systems
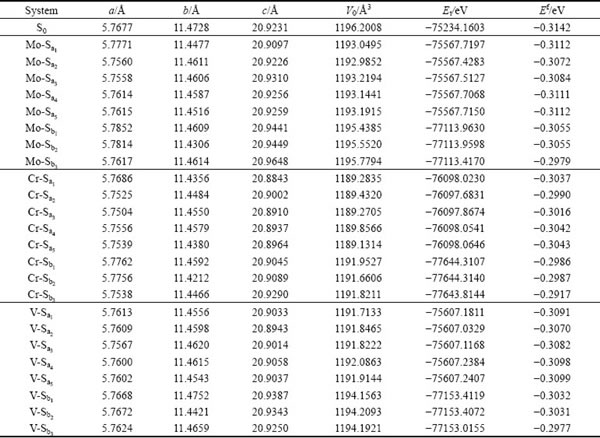
�ɱ�1���Կ�����Mo��Cr��VԪ�طֱ������-TiAl/��2-Ti3Al���洦��Tiԭ�ӻ�Alԭ�Ӻ���������ϵ�ľ�������仯��һ������Mo��Vԭ�ӷֱ����Tiԭ�ӣ�����ϵMo-Sa��V-Sa��a��b��c���н��ͣ���������١���ʹ��ϵ���������仯������Crԭ�����Tiԭ�ӣ�����ϵCr-Sa��a��b��c�����Լ�С��������Լ��١�������Crԭ�ӵĹ��۰뾶(rCr=0.127 nm)����С��Tiԭ�ӵĹ��۰뾶(rTi=0.136 nm)��ռ��Tiλ��Crԭ�Ӿ��нϴ�Ŀ��ƶ��ԡ���������λ�����ƣ����������ڸ��������ԡ�
��Moԭ�����Alԭ�ӣ�����ϵMo-Sb��a��c�����������ӣ�b���н��ͣ���Vԭ�����Alԭ�ӣ�����ϵV-Sb��a���н��ͣ�b��c�������ӡ��ܵ�˵������������ϵ������������͡��ͷų�һ��������������ʹ��ϵ�����������͡�������ϵCr-Sb��a���ӣ�b��c����С���������c�᷽���ԭ�Ӽ���С�������������ڽ��洦�Ľ�ϣ���ϵ��������н��͡��ͷų�һ�����������Ӷ�ʹ����ϵ�����������͡�
���ϵ������ȶ��Կ�����ƽ���γ��ܱ�����һ������£�ԭ��ƽ���γ���Խ�ͣ��ò��ϵ������ȶ���Խ��[32]������Mo��Cr��V����˫���-TiAl/��2-Ti3Al������ϵ��ƽ���γ���Ef���㹫ʽ���Ա�ʾΪ
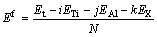 (1)
(1)
ʽ�У�Et��ʾ�Ż�������������ETi��EAl��EX�ֱ��ʾTiԭ�ӣ�Alԭ�ӺͲ���Ԫ��(Mo��Cr��V)�ڵ��ʽṹ��ȫ��ԥ״̬�µĵ�ԭ��������i��j��k�ֱ��ʾ����Ԫ���ھ����е�ԭ������N��ʾ�����е���ԭ�����������㣬�õ����������Ti��Al��Mo��Cr��Vԭ�ӵĵ�ԭ�������ֱ�Ϊ-1603.1192��-56.4638��-1936.8905��-2467.7351��-1976.5062 eV���Ż������������ϵ�����������γ������1���С�
�ӱ�1�п��Կ���������������ϵ��ԭ��ƽ���γ��ܾ�Ϊ��ֵ���������Ǿ����нϺõ������ȶ��ԣ�����һ��ʵ�������²���˫�������ϵ�ǿ�����ʵ���Ʊ������ȶ����ڵġ��������������Mo��Cr��Vԭ�ӷֱ����Tiԭ�Ӻ�Alԭ���γɵ���ϵ��������ϵ��ƽ���γ���Ef����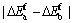 ��0.014 eV����
��0.014 eV����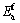 ������
����С��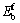 ����ͱ���Cr��Vԭ����λ���ӽ�����ϵʱ������ռ��Tiλ��ռ��Alλ�ĸ��ʲ����һ���ۿ���WEI��[24]�о�������֤����MoԪ����λ���ӵĽ�����ϵ��Moԭ��ռ��Tiλ��ռ��Alλ���ȶ�����Moԭ�������������Tiԭ�ӡ���һ���ۿ����������[19]��HAO��[23]���о�����������֤������Ҫ˵�����ǣ��������[19]�о����ǵ�Ԫ�ز��ӵĦ�-TiAl�ࣻHAO��[23]�о����Ǧ�2-Ti3Al�ࡣ���ڵ�Ԫ�ز��ӵĦ�-TiAl��ͦ�2-Ti3Al�࣬Moԭ�����������Tiԭ�ӵ�ԭ����Ҫ����Moԭ�ӵĹ��۰뾶(rMo=0.145 nm)��Tiԭ�ӵĹ��۰뾶(rTi=0.136 nm)�ȽϽӽ�������Alԭ�ӵĹ��۰뾶(rAl=0.118 nm)��öࡣ��ʹ��Mo���Tiԭ�Ӻ�����ľ������С���෴��Mo���Alԭ�Ӻ������ϴ�ľ�����䣬�����ϵ�ľ��������ߡ��ȶ��Խ��͡�����Moԭ�Ӳ���˫��TiAl/Ti3Al��ϵ����-TiAl�����2-Ti3Al���γɹ������ȷǦ�-TiAl�ࡢҲ�Ǧ�2-Ti3Al�࣬Moԭ��ǡ�þ�������������Ti��Al�����������Mo����˫���-TiAl/��2-Ti3Al��ϵ�����������С��Moԭ��ռλ��������֮�����˺ܴ�仯��������Moռ��Tiλ��ռ��Alλ�ĸ��ʱȽϽӽ�����Զ��ԣ�Cr��Vԭ�ӵĹ��۰뾶(rCr=0.127 nm��rV=0.125 nm)��Tiԭ�Ӻ�Alԭ����ȱ仯���Ǻܴ�����Ļ���̶ȱ仯�����ر���ȶ������������Cr��Vԭ�����Tiλ��Alλ�ĸ��������Moԭ�����Tiλ�ĸ�������һ�㣬���ǵ�ͨ��ʵ�����Ԫ�ز���ʱ���Ͻ�Ԫ���������Ԫ�ض���һ���ĸ��ʡ�
����ͱ���Cr��Vԭ����λ���ӽ�����ϵʱ������ռ��Tiλ��ռ��Alλ�ĸ��ʲ����һ���ۿ���WEI��[24]�о�������֤����MoԪ����λ���ӵĽ�����ϵ��Moԭ��ռ��Tiλ��ռ��Alλ���ȶ�����Moԭ�������������Tiԭ�ӡ���һ���ۿ����������[19]��HAO��[23]���о�����������֤������Ҫ˵�����ǣ��������[19]�о����ǵ�Ԫ�ز��ӵĦ�-TiAl�ࣻHAO��[23]�о����Ǧ�2-Ti3Al�ࡣ���ڵ�Ԫ�ز��ӵĦ�-TiAl��ͦ�2-Ti3Al�࣬Moԭ�����������Tiԭ�ӵ�ԭ����Ҫ����Moԭ�ӵĹ��۰뾶(rMo=0.145 nm)��Tiԭ�ӵĹ��۰뾶(rTi=0.136 nm)�ȽϽӽ�������Alԭ�ӵĹ��۰뾶(rAl=0.118 nm)��öࡣ��ʹ��Mo���Tiԭ�Ӻ�����ľ������С���෴��Mo���Alԭ�Ӻ������ϴ�ľ�����䣬�����ϵ�ľ��������ߡ��ȶ��Խ��͡�����Moԭ�Ӳ���˫��TiAl/Ti3Al��ϵ����-TiAl�����2-Ti3Al���γɹ������ȷǦ�-TiAl�ࡢҲ�Ǧ�2-Ti3Al�࣬Moԭ��ǡ�þ�������������Ti��Al�����������Mo����˫���-TiAl/��2-Ti3Al��ϵ�����������С��Moԭ��ռλ��������֮�����˺ܴ�仯��������Moռ��Tiλ��ռ��Alλ�ĸ��ʱȽϽӽ�����Զ��ԣ�Cr��Vԭ�ӵĹ��۰뾶(rCr=0.127 nm��rV=0.125 nm)��Tiԭ�Ӻ�Alԭ����ȱ仯���Ǻܴ�����Ļ���̶ȱ仯�����ر���ȶ������������Cr��Vԭ�����Tiλ��Alλ�ĸ��������Moԭ�����Tiλ�ĸ�������һ�㣬���ǵ�ͨ��ʵ�����Ԫ�ز���ʱ���Ͻ�Ԫ���������Ԫ�ض���һ���ĸ��ʡ�
Ϊ�˱��ڷ�������������ԭ�Ӳ���˫���-TiAl/��2- Ti3Al��ϵ��ƽ���γ��ܻ���ͼ3�У����ݸ�����ϵ���γ��ܲ���ȷ�����ȶ��IJ���λ�á���ͼ3���Կ�������Mo��Cr��V���Tiԭ���γɵ���ϵ�У���ϵSa5�������ϵͣ�����˵a5�DZȽ��ȶ���λ�á�ͬ������Mo��Cr��V������Alԭ�ӵĸ�����ϵ�У���ϵSb1�������ϵͣ�����˵b1�DZȽ��ȶ���λ�á���ˣ������о���Ҫ����ϵSa5��Sb1Ϊ����
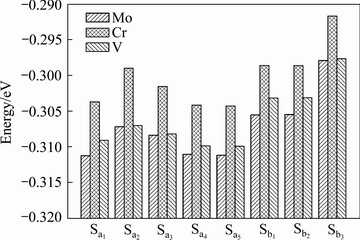
ͼ3 Mo��Cr��V���Ӧ�-TiAl/��2-Ti3Al������ϵ��ƽ���γ���
Fig. 3 Average formation energies of Mo, Cr and V-doping ��-TiAl/��2-Ti3Al interface systems
2.2 ���ӶԽ�����ǿ�ȼ����Ե�Ӱ��
����TiAl���Ͻ������Ǵ������ѻ��ǽ������ѣ���������չ����Ȼ������-TiAl/��2-Ti3Al����档��������ǿ���㹻��ʱ�����ܹ���ֹ���Ƶ���չ������Ͳ�����ֹ����ˣ���һ���̶��������Ľ��ǿ�ȿ����ö��ѹ��������������������ϵ��Griffith���ѹ�W[33]�ɱ�ʾΪ
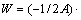
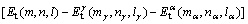 (2)
(2)
ʽ�У�AΪ����ģ�͵ĺ�������Et (m, n, l)Ϊ�Ż����-TiAl/��2-Ti3Al�������ϵ����������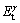 (m��, n��, l��)��
(m��, n��, l��)��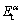 (m��, n��, l��)�ֱ�Ϊ�γɽ���ǰ��-TiAl (111)��ͦ�2-Ti3Al (0001)�����������m��n��l�ֱ�ΪTi��Al�Ͳ���Ԫ��X��ԭ�Ӹ�������m=m��+m����n=n��+n����l=l��+l����
(m��, n��, l��)�ֱ�Ϊ�γɽ���ǰ��-TiAl (111)��ͦ�2-Ti3Al (0001)�����������m��n��l�ֱ�ΪTi��Al�Ͳ���Ԫ��X��ԭ�Ӹ�������m=m��+m����n=n��+n����l=l��+l����
�ɱ�2�еľ�������ɵã���ϵS0��Mo-Sa5��Cr-Sa5��V-Sa5��Mo-Sb1��Cr-Sb1��V-Sb1������������ֱ�Ϊ0.6617��0.6598��0.6581��0.6598��0.6630��0.6619��0.6618 nm3�������Ǵ���ʽ(2)������õ���Ӧ�Ķ��ѹ��ֱ�Ϊ3.22��3.18��3.19��3.22��3.27��3.26��3.28 J/m2��Ϊ���ڱȽϣ�������ϵ�Ķ��ѹ�����ͼ4�С�����ϵS0��Ƚϣ���ϵMo-Sa5��Cr-Sa5�Ķ��ѹ����в�ͬ�̶ȵ��½�������������ڽ��洦�IJ���ԭ�ӻᵼ�½��ǿ�Ƚ��͡�����ϵV-Sa5�Ķ��ѹ��������䣬����Vռ��Tiλʱ���Խ���Ľ��ǿ��Ӱ���С����ϵMo-Sb1��Cr-Sb1��V-Sb1�Ķ��ѹ����в�ͬ�̶ȵ����ߡ�����������߽���Ľ��ǿ�ȡ���һ���ۿ���κǿ��[20]�о�������֤�����Ͽ�֪����ϵMo-Sa5��Cr-Sa5�Ľ��ǿ�ȼ����������ڸ��ƺϽ���ϵ����ԣ���ϵV-Sa5�Ľ��ǿ�ȱ仯�������Ը��ƹ���ҲС����ϵMo-Sb1��Cr-Sb1��V-Sb1�Ľ��ǿ���������ڸ��ƺϽ���ϵ����ԡ�
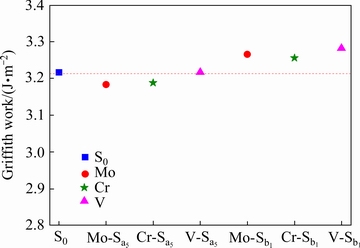
ͼ4 Mo��Cr��V���Ӧ�-TiAl/��2-Ti3Al������ϵ�Ķ��ѹ�
Fig. 4 Griffith works of Mo, Cr and V-doping ��-TiAl/��2-Ti3Al interface systems
2.3 �����Բ�����ϵ��̬�ܶ�����ǿ��
Ϊ�˽�һ�������˽�Mo��Cr��VԪ�ز��ӶԦ�-TiAl/��2-Ti3Al�Ͻ�������Ը��Ƶ��ۻ��ƣ�ѡȡ���ѹ���С�����С�����������ϵMo-Sa5��V-Sa5��V-Sb1�Լ���ϵS0����������������洦�ĵ���̬�ܶ�(DOS)�ͷֲ�̬�ܶ�(PDOS)����ͼ5��ʾ���ݴ˽��жԱȷ�����
��ͼ5����̬�ܶ�ͼ���Կ�����������ϵ�ڷ����ܼ���������Ũ�Ȳ�Ϊ�㡣��˵������ǰ��ĸ�����ϵ���������ԵĽ������ʡ�������ϵS0����̬�ܶ��ڿ��������ܼ����������������壬��M0���N0�塣�ɼ����ӵ�����������Ҫ�ֲ���-9.59~1.0 eV���ڴ��������䣬Tiԭ�ӵ�3d���Ӻ�Alԭ�ӵ�3p������ص������ӹ��������ǿ���ӻ����á�Ti��Alԭ�ӵķֲ�̬�ܶ���ʾ��M0����Ҫ��Ti-d���Ӻ�Al-p���ӹ��ף�����ҪΪp-d����ӻ��塣�����Tiԭ�Ӻ�Alԭ��֮����к�ǿ�Ĺ��ۼ���ϣ��Ӷ�ʹTiAl�Ͻ�����ȶ��Ľṹ������Morinaga����[22]��Ti-d������Al-p�����γɵĹ��ۼ����н�ǿ�ķ����ԡ����ǵ���TiAl�Ͻ���Եĸ���ԭ��N0����Ҫ�ɴ���Ti-d���Ӻ�����Al-p���ӹ��ף�����ҪΪTi��Ti����ͬʱ����������Ti��Al����������ϵMo-Sa5���ɼ�����������Ҫ�ֲ���-9.61~1.0 eV��������̬�ܶ�ͼ��Ҳ�����������ɼ��壬��M1���N1�塣������Ҫ��Ti-3d���ӣ�Al-3p���Ӻ�Mo-4d���ӹ��ס��Ա�ͼ5(b)��(a)��֪����ϵMo-Sa5��p-d����ӻ��ɼ���(M1, 24.22 eV-1)������ϵS0(M0, 25.14 eV-1)������ϵMo-Sa5��p-d����ӻ���ǿ�ȵ�����ϵS0������ζ��Mo���Ӻ�ʹ����p-d����ӻ��ĵ��������٣������˹��ۼ�ǿ�ȡ���Ӧ�أ�λ���˶����������٣����������Եĸ��ơ���һ������ʹHUANG��[34]����Ti-Al-Mo��Ԫ��ϵ��ʵ�������˶��������ݡ����ǵ�ʵ���о�����������һ������MoԪ�أ��ܹ��ı�TiAl���Ͻ��Ц���ͦ�2����ȶ��ԣ�ʹTi-Al-Mo��Ԫ��ϵ�ﵽƽ�⡣������ϵV-Sa5���ɼ����ӵ�����������Ҫ�ֲ���-9.59~1.0 eV��������Ҫ��Ti-3d���ӡ�Al-3p�����Լ�V-3d���ӹ��ס��Ա�ͼ5(c)��(a)��֪����ϵV-Sa5��p-d����ӻ��ɼ���(M2��24.83 eV-1)������ϵS0��Ҳ����˵��ϵV-Sa5����ϵS0��p-d����ӻ���ǿ���Ե͡�����ζ�Ų�������V����ʹ����p-d����ӻ��ĵ��������м��٣��Խ���Ľ��ǿ��Ӱ���С��������ϵV-Sb1���Ա�ͼ5(d)��(a)��֪���ɼ���M3��N3�ص�����ϵV-Sb1��p-d����ӻ��ɼ���(M3, 19.45 eV-1)���Ե�����ϵS0��p-d����ӻ����壻ͬʱN3��Ҳ���Խ��͡���ˣ�������p-d����ӻ����彵�Ͳ���Ԥ��۽�ϼ�����ԭ�����γ�Ti��Al��Ti��Ti��Al��Al���ĵ��������ڼ��١�Ȼ���ۺ�ͼ5�ɼ���������ϵS0��Mo-Sa5��V-Sa5��Al-p������Ti-d���ӵĹ���ӻ��ɼ��������Ե���Ti-d��Ti(Mo,V)-d���ӵĹ���ӻ��ɼ��壻����ϵV-Sb1��p������d���ӵĹ���ӻ��ɼ����Ѿ���Ti(V)-d���ӹ���ӻ������죬��Զ��ԣ�ʵ����p-d����ӻ��ĵ���̬�ܶȴ���������������Գ̶���ǿ�����ߣ��ڷ����ܼ�������ϵV-Sb1�ĵ���̬�ܶ�(15.74 eV-1)���Ե�����ϵS0��(22.35 eV-1)��Ҳ����V���Alԭ�Ӻ�ʹ��ϵ�����������ͣ������˽���Ľ��ǿ�ȣ������ڸ�����ϵ�����ԡ�
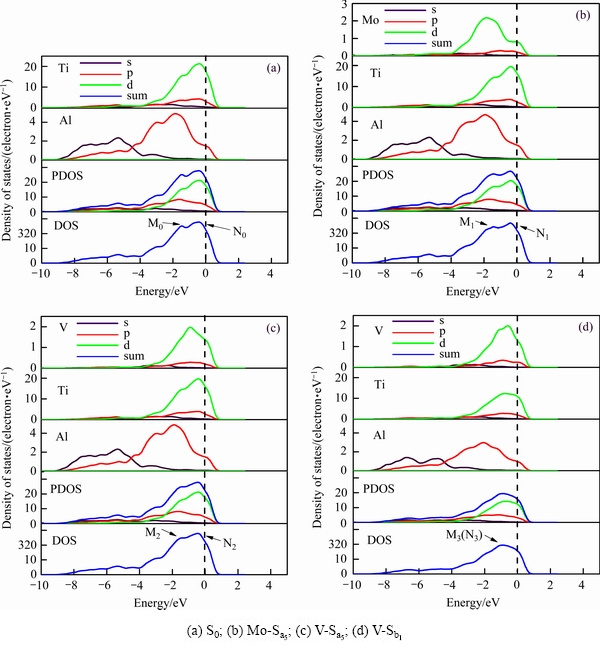
ͼ5 ������ϵ�ķ����ܼ������ĵ���̬�ܶ�
Fig. 5 Densities of states of interface systems near Fermi level
2.4 �����Բ�����ϵ�ĵ���ܶ��뻯ѧ������
��Ȼ����Mo��V���Ӻ�����������Ti��Alԭ���γɽ����������Ӽ������Ͻ��й��ۼ�Ti��Ti��Al��Al��Al��Ti��ռ����Ҫ����[35]��Ϊ�˽�һ���о�Mo��V���Ӻ�����Χԭ�ӵļ������ã�ͼ6�ֱ��������ϵS0��Mo-Sa5��V-Sa5��V-Sb1�ľ��д����Ե�(001)��ĵ���ܶȡ�

ͼ6 ������ϵS0��Mo-Sa5��V-Sa5��V-Sb1������ԭ������(001)��ĵ���ܶȷֲ�
Fig. 6 Maps of charge density of crystal face (001) passing through impurity atom in interface systems
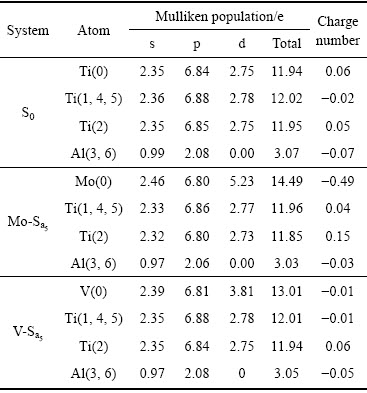
��ͼ6���Կ���������ϵS0�У�Tiԭ����Alԭ��ͨ�����õ��Ӷ��γɹ��ۼ������н�ǿ�ķ����ԡ��Ա�ͼ6(b)��(a)���Կ���������ϵMo-Sa5�У�����ԭ��Mo��Χ�ĵ���ܶ��������ӡ���(001)���ϣ�Moԭ�����ĸ����ڵ�Tiԭ��֮��ĵ����ơ�������չ��ǿ�����ӡ����������Mo���Tiԭ�Ӻ�Mo-d������Ti-d�����ӻ�������ǿ�����Ƶأ�Moԭ�����������ڵ�Alԭ��֮��ĵ����ƣ�Ҳ���С�������չ��ǿ�����ӡ����ص㡣��Ҳ����Mo-d������Al-p���ӵ��ӻ�������ǿ������ϸ�۲죬������Alԭ����Χ��ɫ�ĵ��ܶȵ�������״�����˱仯������ϵS0������״Ϊ�������Ρ���������ϵMo-Sa5������״���ӽ���Բ�Ρ�����Ҳ����˵������ϵMo-Sa5���ԣ�Alԭ����Χ����ͬ�Գ̶�����ˣ����Ӽ���������������ǿ�ˡ�Ϊ��ʹ��һ�жϸ���˵��������2������(001)����λ�á�0������Tiԭ��(������Mo��Vԭ��)��������ڵ�����ԭ�ӵ�Mulliken����������3������λ�á�0������Tiԭ��(������Mo��Vԭ��)��������ڵ�����ԭ��֮����ص�����������Mulliken��������֪������ϵS0�н��洦Ti(0)ԭ�ӵ��ܵ����Ϊ0.06������ϵMo-Sa5��Moԭ�ӵ��ܵ��������Ϊ-0.49�������Moȷʵ����˵��ӡ�Al(3, 6)�ĵ������-0.07��Ϊ-0.03�����������s-p����ӻ��ĵ��������١����ص���������֪��Mo���Tiԭ�Ӳ��Ӻ�Mo��Alԭ��֮����ص�������Ϊ0.17��������ϵS0��Ti��Alԭ��֮����ص�������0.20�������Mo��Alԭ�Ӽ�Ĺ������Լ������ۺ϶��ԣ�Mo�IJ��������Moԭ��ʵ��������ʹ���ڽ���������Ʒֲ��������Եľۼ�ЧӦ������Ϊ��Χ��Tiԭ�Ӻ˺�Alԭ��ʵ�����磬����Moԭ��ʵ���������ã����ﵽƽ�⡣�ۺ�Ч�����ǣ���Moԭ�ӵ���Χ�γɽ��ǿ���Ըߵ�������������Χ��������Ľ�ϵĸ������Գ̶��½����������ڸ��Ʋ��ϵ����ԡ��������������MANDA��[9]��ʵ��������Ǻϡ��Ա�ͼ6(c)��(a)���Է��֣�����ϵV-Sa5��Vԭ����Χ�������ϵMo-Sa5��Moԭ����Χ������ƣ������С�������չ��ǿ�����ӡ����ص㣬���̶��Ե͡���һ�жϵõ�ԭ�ӵ�������ص���������֧�֡���ˣ��ۺ�Ч��Ҳ����Vԭ�ӵ���Χ�γɽ��ǿ���Ըߵ�������������Χ������������õĸ������Գ̶��½����������ڸ��Ʋ��ϵ����ԡ��Ա�ͼ6(d)��(a)���֣�����ϵV-Sb1��Vԭ�Ӳ����ƺ����������෴�ķ���չ�����ƣ��������Ƶķ��ۼ�ЧӦ����¸���������Χ������������õĸ������Գ̶���������Ͳ����ڸ��Ʋ��ϵ����ԡ�
��2 ��ϵS0��Mo-Sa5��V-Sa5��ԭ�ӵ�Mulliken�������͵����
Table 2 Mulliken populations and charges numbers in system S0, Mo-Sa5 and V-Sa5
��3 ��ϵS0��Mo-Sa5��V-Sa5�е��ص�������
Table 3 Overlap populations of system S0, Mo-Sa5 and V-Sa5.

3 ����
1) Mo��Cr��VԪ�ع�����˫���-TiAl/��2-Ti3Al�������������ϵ����������ƽ���γ��ܾ�Ϊ��ֵ���������ǿ�����ʵ���Ʊ������ȶ����ڡ�����Mo��Cr��VԪ�����Ti��Alԭ�Ӻ�����ϵ��ƽ���γ������������ռ��Tiλ��ռ��Alλ�ĸ��ʲ��
2) ���ѹ���ʾ����ϵMo-Sa5��Cr-Sa5�Ľ��ǿ�ȼ����������ڸ��ƺϽ���ϵ����ԣ���ϵV-Sa5�Ľ��ǿ�ȱ仯�������Ը��ƹ���ҲС����ϵMo-Sb1��Cr-Sb1��V-Sb1�Ľ��ǿ���������ڸ��ƺϽ���ϵ����ԡ�
3) ̬�ܶ���ʾ������ϵMo-Sa5 (��V-Sa5)��Mo(��V)Ԫ�����Tiԭ�Ӳ���ʹ����p-d����ӻ��ĵ��������٣����ǿ�Ƚ��ͣ�������λ���˶����谭�����������ڸ��Ʋ��ϵ����ԣ���VԪ�ز��ӵ�Ӱ�����������ϵV-Sb1��V���Alԭ�Ӻ�ʹ��ϵ�����������ͣ������˽���Ľ��ǿ�ȣ������ڸ��Ʋ��ϵ����ԡ�
4) ����ܶ���ʾ������ϵMo-Sa5(��V-Sa5)��Mo(��V)�IJ���ʹ����Χ�γɽ��ǿ���Ըߵ�������������Χ��������Ľ�ϵĸ������Գ̶��½����������ڸ��Ʋ��ϵ����ԡ�������ϵV-Sb1�У�Vԭ�Ӳ�������������Ƶķ��ۼ�ЧӦ�����ƣ���Ͳ����ڸ��Ʋ��ϵ����ԡ�
REFERENCES
[1] CLEMENS H, MAYER S. Design, processing, microstructure, properties, and applications of advanced intermetallic TiAl alloys [J]. Advanced Engineering Materials, 2013, 15(4): 191-215.
[2] BEWLAY B P, NAG S, SUZUKI A, WEIMER M J. TiAl alloys in commercial aircraft engines[J]. Materials at High Temperatures, 2016, 33 (4/5): 549-559.
[3] WU X H. Review of alloy and process development of TiAl alloys[J]. Intermetallics, 2006, 14(10/11): 1114-1122.
[4] KANANI M, HARTMAIER A, JANISCH R. Stacking fault based analysis of shear mechanisms at interfaces in lamellar TiAl alloys[J]. Acta Materialia, 2016, 106: 208-218.
[5] SHU S L, TONG C Z, QIU F, ZOU Q, JING Q C. Effects of ternary elements on the ductility of TiAl[J]. The Canadian Journal of Metallurgy and Materials Science, 2016, 55(2):156-160.
[6] �Ž�ΰ, ������, ������, ���ƾ�. Ti3A1��Ti2A1Nb���Ͻ���о���Ӧ��[J]. �й���ɫ����ѧ��, 2010, 20(1): 336-341.
ZHANG Jian-wei, LI Shi-qiong, LIANG Xiao-bo, CHENG Yun-jun. Research and application of Ti3A1 and Ti2A1Nb based alloys[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(1): 336-341.
[7] SASTRY S M L, LIPSITT H A. Fatigue deformation of TiAl base alloys[J]. Metallurgical and Materials Transactions A, 1997, 8(2): 299-308.
[8] OEHRING M, APPEL F, ENNIS P J, WAGNER R, A TEM study of deformation processes and microstructural changes during long-term tension creep of a two-phase ��-titanium aluminide alloy[J]. Intermetallics, 1999, 7(3/4): 335-345.
[9] MANDA P, PATHAK A, MUKHOPADHYAY A, CHAKKINGAL U, SINGH A K. Ti-5Al-5Mo-5V-3Cr and similar Mo equivalent alloys: First principles calculations and experimental investigations[J]. Journal of Applied Research and Technology, 2017, 15: 21-26.
[10] XI Y J, LIU Y J, WANG Z X, LIU J B. Oxidation and electrochemical corrosion performance of Ti3Al alloy with TiAl coating[J]. Anti-Corrosion Methods and Materials, 2010, 57(1): 13-17.
[11] �� ��. ���������仯����Ľ�չ����ս[J]. ����ѧ��, 2015, 51(2): 129-147.
YANG Rui. Advances and challenges of TiAl base alloys[J]. Acta Metallurgica Sinica, 2015, 51(2): 129-147.
[12] HU H, WU X Z, WANG R, LI W G, LIU Q. Phase stability, mechanical properties and electronic structure of TiAl alloying with W, Mo, Sc and Yb: First-principles study[J]. Journal of Alloys and Compounds, 2016, 658: 689-696.
[13] ���칦, ���, �Կ���, �ع�˳, ������, ��ѩ��. Zr��(��)Mn��λ���Ӧ�-TiAl���Ͻ���������������[J]. �й���ɫ����ѧ��, 2016, 26(11): 2309-2318.
SONG Qing-gong, YANG Bao-bao, ZHAO Jun-pu, QIN Guo-shun, GUO Yan-rui, HU Xun-lan. Investigations on ductibility and electronic property of Zr and (or) Mn doped ��-TiAl based alloys[J]. The Chinese Journal of Nonferrous Metals, 2016, 26(11): 2309-2318.
[14] ������, ������, Ф����, ������, �� ��. ϡ��Y�ڦ�-TiAl���Ͻ��侫���ȳ�����Ӧ�õ��о���չ[J]. �й���ɫ����ѧ��, 2014, 24(5): 1241-1251.
CHEN Yu-yong, HAN Jian-chao, XIAO Shu-long, XU Li-juan, TIAN Jing. Research progress of rare earth yttrium application in ��-TiAI based alloy and precision thermal forming[J]. The Chinese Journal of Nonferrous Metals, 2014, 24(5): 1241-1251.
[15] WANG L, SHANG J X, WANG F H, ZHANG Y. First principles study of ��2-Ti3Al (0001) surface and/��-TiAl(111)/��2-Ti3Al (0001) interfaces[J]. Applied Surface Science, 2013, 276: 198-202.
[16] ZGHAL S, NAKA S, COURET A, A quantitative TEM analysis of the lamellar microstructure in TiAl based alloys[J]. Acta Materialia, 1997, 45(7): 3005-3015.
[17] ףӨӨ, �� ��, ��˳ƽ, ������, ������, ������, ������. ����������û�����о�Cr��Nb��TiAl�Ͻ���ȱ�ݺ�d-d��������õ�Ӱ��[J]. ϡ�н��������빤��, 2009, 38(2): 271-274.
ZHU Ying-ying, DENG Wen, SUN Shun-ping, JIANG Hai-feng, HUANG Yu-yang, CAO Ming-zhou, XIONG Liang-yue. Influence of Cr and Nb on defects and d-d electron interactions in TiAl alloys researched by positron annihilation techniques[J]. Rare Metal Materials and Engineering, 2009, 38(2): 271-274.
[18] �� ��, �Ʋ���, �׳���, ������. ���Ӹ�Ԫ�ض�TiAl�Ͻ��������Ժ��۽ṹ��Ӱ��[J]. ���Ͽ�ұѧԺѧ��, 1992, 23(5): 560-564.
XIONG Xiang, HUANG Bai-yun, LEI Chang-ming, L�� Hai-bo. Effect of Cr addition on room-temperature ductility and microstructure of TiAl alloy[J]. Journal of Central South Institute of Mining and Metallurgy, 1992, 23(5): 560-564.
[19] ������, ��ǰ��, ������, ������. ����Ԫ�ز��Ӷ�TiAl�Ͻ���ѧ���ܵ�Ӱ��[J]. ����ѧ��, 2016, 65(7): 1-9.
WANG Hai-yan, HU Qian-ku, YANG Wen-peng, LI Xu-Sheng. Influence of metal element doping on the mechanical properties of TiAl alloy [J]. Acta Physica Sinica, 2016, 65(7): 1-9.
[20] κ ǿ, �� ˶. ������ԪCr, Mo, W���Ӷ�������������Ե�Ӱ��[J]. �������ܲ���, 2014, 21(2): 26-31.
WEI Qing, WANG Shuo. Effect on brittle of titanium aluminum compound by doping Cr, Mo, W additions[J]. Metallic Functional Materials, 2014, 21(2): 26-31.
[21] ������, ������, �� ��. ��-TiAl��Nb��Mo�Ͻ�ЧӦ�ĵ�һ��ԭ���о�[J]. ����ѧ��, 2007, 56(5): 2838-2844.
DANG Hong-li, WANG Chong-yu, YU Tao. First-principles investigation on alloying effect of Nb and Mo in ��-TiAl[J]. Acta Physica Sinica, 2007, 56(5): 2838-2844.
[22] MORINAGA M, SAITO J, YUKAWA N, ADACHI H. Electronic effect on the ductility of alloyed TiAl compound[J]. Acta Metallurgica et Materialia, 1990, 38(1): 25-29.
[23] HAO Y L, XU D S, CUI Y Y, YANG R, LI D. The site occupancies of alloying elements in TiAl and Ti3Al alloys[J]. Acta Materialia, 1999, 47(4): 1129-1139.
[24] WEI Y, ZHANG Y, LU G H, XU H B. Effects of transition metals in a binary-phase TiAl-Ti3Al alloy: From site occupancy, interfacial energetics to mechanical properties[J]. Intermetallics, 2012, 31: 105-113.
[25] �� ��, ��ΰ��, �� ��, ��־��. Ti3Al���Ͻ�(0001)��2//(110)������۵��ӽṹ����[J]. ϡ�н��������빤��, 2005, 34(10): 1569-1573.
QU Hua, LIU Wei-dong, ZHANG Kun, LIU Zhi-lin. Study on the valence electron structure of (0001)��2//(110)�� interface in Ti3Al-based alloys[J]. Rare Metal Materials and Engineering, 2005, 34(10): 1569-1573.
[26] ��ΰ��, �� ��, ��־��. ˫��TiAl�Ͻ��2/�ý�����ӽṹ���������ͻ��Ʒ���[J]. ϡ�н��������빤��, 2005, 34(2): 199-204.
LIU Wei-dong, QU Hua, LIU Zhi-lin. Calculation of valence electron structures of ��2/�� interface and toughening mechanism in two-phase TiAl-alloy[J]. Rare metal materials and engineering, 2005, 34(2): 199-204.
[27] WEI Y, ZHANG Y, ZHOU H B, LU G H, XU H B. First-principles investigation on shear deformation of a TiAl/Ti3Al interface and effects of oxygen[J]. Intermetallics, 2012, 22: 41-46.
[28] FISCHER F D, WAITZ T, SCHEU C, CHA L, DEHM G, ANTRETTER T, CLEMENS H. Study of nanometer-scaled lamellar microstructure in a Ti-45Al-7.5Nb alloy-experiments and modeling[J]. Intermetallics, 2010, 18: 509-517.
[29] KOIZUMI Y, FUJITA T, MINAMINO Y, HATA S. Effects of plastic deformation on lamellar structure formation in Ti-39 at.% Al single crystals[J]. Acta Materialia, 2010, 58: 1104-1115.
[30] ��Ѽ�, ������, �� ��, ������. Cu��Ni ����FeS2���ӽṹ���ѧ���ʵĵ�һ��ԭ������[J]. �й���ɫ����ѧ��, 2017, 27(3): 605-612.
WU Jia-jia, MA Wan-kun, JIAO Fen, QIN Wen-qing. First principle calculation of electronic structures and optical properties of copper and nickel doped FeS2[J]. The Chinese Journal of Nonferrous Metals, 2017, 27(3): 605-612.
[31] �� ��, ������. �����Ѿ�������ɢ��Ϊ�ĵ�һ��ԭ��[J]. �й���ɫ����ѧ��, 2015, 25(11): 3100-3106.
LIU Song, WANG Yin-gang. First-principles of hydrogen diffusion mechanism in titanium crystals[J]. The Chinese Journal of Nonferrous Metals, 2015, 25(11): 3100-3106.
[32] ������, �� ��, ��Т��, ��Ч˫, ½ ��. ���ӶԽ���-MoS2�������ʵ��Ƶĵ�һ��ԭ���о�[J]. ����ѧ��, 2017, 66(11): 11820.
TAO Peng-cheng, HUANG Yan, ZHOU Xiao-hao, CHEN Xiao-shuang, LU Wei. First principles investigation of the tuning in metal-MoS2 interface induced by doping[J]. Acta Physica Sinica, 2017, 66(11): 11820/1-11820/8.
[33] ��С��, �Ų���, ����Ƽ, �����. Al��Zn ��Mg-Li���Ͻ�ЧӦ�ĵ�һ��ԭ���о�[J]. ϡ�н��������빤��, 2014, 43(7): 1661-1665.
WANG Xiao-hong, ZHANG Cai-li, HU Xiu-ping, HAN Pei-de. First principle study on alloying effect of Al and Zn doping on Mg-Li phase interface[J]. Rare Metal Materials and Engineering, 2014, 43(7): 1661-1665.
[34] HUANG X M, ZHU L L, CAI G M, LIU H S, JIN Z P. Experimental investigation of phase equilibria in the Ti�CAl�CMo ternary system[J]. Journal of Materials Science, 2017, 52: 2270-2284.
[35] ���칦, �Կ���, ������, �絤��, ������, ������. �����ܶȷ������۵�La���Ӧ�-TiAl��ϵ�ṹ�������������[J]. ����ѧ��, 2017, 66(6): 066103.
SONG Qing-gong, ZHAO Jun-pu, GU Wei-feng, ZHEN Dan-dan, GUO Yan-rui, LI Ze-peng. Ductile and electronic properties of La-doped gamma-TiAl systems based on density functional theory[J]. Acta Physica Sinica, 2017, 66(6): 066103.
Effect of Mo(or Cr, V) substitution doping on energy, ductility and electronic properties of dual-phase ��-TiAl/��2-Ti3Al interface
SONG Qing-gong1, 2, GU Wei-feng1, ZHEN Dan-dan2, GUO Yan-rui1, HU Xue-lan2
(1. Institute of Low Dimensional Materials and Technology, College of Science, Civil Aviation University of China, Tianjin 300300, China;
2. Sino-European Institute of Aviation Engineering, Civil Aviation University of China, Tianjin 300300, China)
Abstract: The average formation energies, Griffith fracture works and electronic structures of Mo (or Cr, V) doping of ��-TiAl/��2-Ti3Al interfacial systems were calculated with density functional theory. The results indicate that these systems possess energy stability and can be prepared by experiments and exist stably. The Griffith fracture work and density of the representative system show that the bonding strength of Mo-Sa5 (or Cr-Sa5) is weakened and the densities of state of Mo-d and Ti-d electrons of Mo-Sa5 are increased. The charge density map of the (001) plane passing through impurity atom and populations of the doping system show that, there is an electron cloud aggregation effect surrounding the dopant caused by the doping of Mo (or V) atom, which forms a region with a slightly higher bonding strength. As a result, the anisotropy degree of the combination of this region is reduced, which is the internal reason of the improved ductility for TiAl alloys.
Key words: dual-phase ��-TiAl/��2-Ti3Al interface; energy property; ductility; electronic property; first-principle
Foundation item: Project(51201181) supported by the National Natural Science Foundation of China; Project (3122016L012) supported by the Fundamental Research Funds for the Central Universities, China
Received date: 2017-08-24; Accepted date: 2017-11-27
Corresponding author: SONG Qing-gong; Tel: +86-22-24092510; E-mail: qgsong@cauc.edu.cn
(�༭ ��ѧ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(51201181)�������У��������ҵ���ר��������Ŀ(3122016L012)
�ո����ڣ�2017-08-24�������ڣ�2017-11-24
ͨ�����ߣ����칦�����ڣ���ʿ���绰��022-24092510��E-mail��qgsong@cauc.edu.cn

