J. Cent. South Univ. (2017) 24: 1703-1712
DOI: https://doi.org/10.1007/s11771-017-3577-7
Electrocatalytic behaviour of Ni and NiCu alloy modified glassy carbon electrode in electro-oxidation of contraflam
Naeemy Ali1, Mohammadi Ali1, 2, Ehsani Ali3, Aghassi Ali1
1. Department of Drug and Food Control and Pharmaceutical Quality Assurance Research Centre, Faculty of
Pharmacy, Tehran University of Medical Sciences, P.O. Box 14155-6451, Tehran, Iran;
2. Nanotechnology Research Centre, Faculty of Pharmacy, Tehran University of Medical Sciences,
P.O. Box 14155-6451, Tehran, Iran;
3. Department of Chemistry, Faculty of Science, University of Qom, P.O. Box 37185-359, Qom, Iran
 Central South University Press and Springer-Verlag GmbH Germany 2017
Central South University Press and Springer-Verlag GmbH Germany 2017
Abstract: The electrocatalytic oxidation of contraflam was investigated in alkaline solution on nickel and nickel–copper alloy modified glassy carbon electrodes (GC/Ni and GC/NiCu). We prepared these electrodes by galvanostatic deposition and the surface morphologies and compositions of electrodes were determined by energy-dispersive X-ray (EDX) and scanning electron microscopy (SEM). Cyclic voltammetry and chronoamperometric methods were employed to characterize the oxidation process and its kinetics. Voltammetric studies exhibit one pair of well-defined redox peaks, which is ascribed to the redox process of the nickel and followed by the greatly enhanced current response of the anodic peak in the presence of contraflam and a decrease in the corresponding cathodic current peak. This indicates that the immobilized redox mediator on the electrode surface was oxidized contraflam via an electrocatalytic mechanism. The catalytic currents increased linearly with the concentration of contraflam in the range of 0.25–1.5 mmol/L. The anodic peak currents were linearly proportional to the square root of scan rate. This behaviour is the characteristic of a diffusion-controlled process. The determination of contraflam in capsules is applied satisfactorily by modified electrode.
Key words: galvanostatic deposition; nickel; nickel-copper alloy; contraflam; modified electrodes; cyclic voltammetry; oxidation
1 Introduction
Contraflam (2-[(2, 3-dimethylphenyl) amino] benzoic acid, Fig. 1), mefenamic acid (MFA), is an important non-steroidal anti-inflammatory drug (NSAID) used to treat several pathologies. It is used to relieve the symptoms of many diseases such as rheumatoid arthritis, osteoarthritis, non-articular rheumatism, and sports injuries [1, 2]. Overdoses of MFA produce toxic metabolite accumulation that causes acute hepatic necrosis, inducing morbidity and mortality in humans [2]. Due to the vital importance of the assay of MFA for pharmaceutical formulations and dosage forms, several analytical methods have been developed for the quantitative determination of the MFA in pharmaceutical samples. These methods include spectrophotometry [3, 4], chromatography [5], chemiluminescence [6], capillary electrophoresis [7, 8], polarography [9, 10], molecularly imprinted polymers [11] and electrochemistry [12, 13].
Chemically modified electrodes, which are of recent interest as chemical sensors, have attracted immense attention because of their sensitivity and selectivity in the analysis of organic compounds in food and drug products [4, 15]. The slow electron transfer kinetics on the bare/ unmodified electrode surface is substantially changed by modifying the bare electrode, which in turn speeds up the electron transfer kinetics of the reaction on the surface and enables the reaction to occur at a faster rate and an appreciable reduced over voltage [16, 17]. It is well established that nickel can be used as a catalyst due to its surface oxidation properties. Ni has commonly been used as an electrocatalyst for both anodic and cathodic reactions (oxidation and determination) of a large number of organics [8-23]. The fact that pure Ni and Cu metals have the same face centred cubic structure with similar lattice parameters (α=3.523  for Ni and 3.616 for Cu) makes it possible to have a wide range of composition for NiCu alloy. Numerous papers have described in chemical and physical properties of Ni–Cu alloys [24, 25]. KHULBE et al [25] presented an excellent review on the behaviour of NiCu alloy in a variety of catalytic mechanisms, including hydrogenation reactions, ortho–para hydrogen conversion, and H2/D2 exchange reaction.
for Ni and 3.616 for Cu) makes it possible to have a wide range of composition for NiCu alloy. Numerous papers have described in chemical and physical properties of Ni–Cu alloys [24, 25]. KHULBE et al [25] presented an excellent review on the behaviour of NiCu alloy in a variety of catalytic mechanisms, including hydrogenation reactions, ortho–para hydrogen conversion, and H2/D2 exchange reaction.
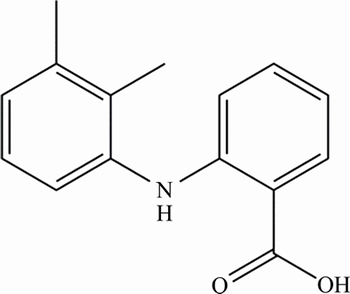
Fig. 1 Chemical structure of MFA
The purpose of the present work is to study the electrochemical oxidation of the MFA on Ni and NiCu alloy modified GC electrodes in a solution of 0.1 mol/L NaOH. The practical application of the modified electrode was illustrated by measuring the amount of MFA concentration in capsules.
2 Experimental
Nickel sulphate, copper sulphate, sodium hydroxide, sodium citrate used in this work were at least of the analytical grade of Merck origin and were used without further purification. All solutions were prepared with double distilled water throughout. The MFA standard powder was obtained as a gift from the Center of Quality Control of Drug, Tehran, Iran.
A stock solution of MFA (0.5 mg/mL) was prepared in 0.1 mol/L NaOH solution. For the preparation of standard solutions, an aliquot of the stock solution of the MFA was transferred to a volumetric flask and made up to volume with 0.1 mol/L NaOH solution. In all cases, the prepared solutions were protected from light using aluminium foil and stored at 4 °C for 48 h.
Electrochemical studies were carried out in a conventional three-electrode cell powered by an electrochemical system comprising of AUTOLAB system with micro III (Eco Chemie BV, Netherlands). The system is run by a PC through Nova commercial software (Eco Chemie); A dual Ag/AgCl–saturated KCl, a platinum rod and GC electrodes (from Metrohm) were used as the reference, counter and working electrodes, respectively. All measurements were carried out at (25±2) °C. The cell was purged with pure nitrogen gas before every measurement for the dissolved oxygen should be removed from the working solution (the measurement must be done in an oxygen-free solution).
The GC disk electrode supplied by AUTOLAB was further polished with 0.05 μm α-alumina powder on a polishing micro cloth. Then, the electrode was placed in a 1:1 distilled water/ HNO3 solution for about 5 min, and followed by placing in an ultrasonic bath with a 1:1 mixture of acetone/water for another 5 min and then rinsed thoroughly with double distilled water prior to modification.
Films of nickel were formed on the GC surface by galvanostatic deposition. The current density was 10 mA/cm2 for 300 s while the electrode substrate was immersed in the middle of the N2 purged mixture of 0.7 mol/L NiSO4·6H2O +0.26mol/L C6H5Na3O7·2H2O solution for Ni deposition and the addition of 0.05 mol/L CuSO4·5H2O for NiCu alloy deposition (electrolyte was stirred with a magnetic stirrer during electrodeposition). SEM (Philips XL30, USA) used to evaluate the chemical composition of the deposit and equipped with EDX.
3 Results and discussion
The morphology and chemical composition analysis of prepared composite was characterized using SEM and EDX. Figure 2 depicts the EDX spectrum, which shows the chemical composition analysis of the NiCu composite that contains 79 % Ni and 21 % Cu.
The SEM micrographs presented in Fig. 3 display the morphology of the bare GC and electrodeposited Ni and NiCu alloy obtained by SEM. Figure 3 clearly shows well-exfoliated, wrinkled 2D NiCu alloy with the higher surface area rather than only Ni on the GC electrode surface.
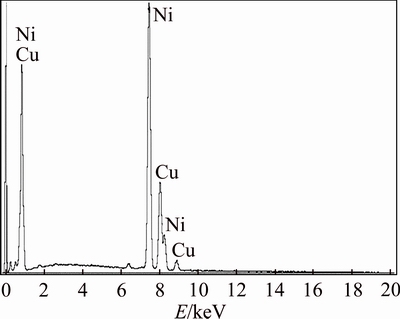
Fig. 2 EDX results of chemical composition of NiCu alloy on surface of GC
Figure 4 illustrates successive cyclic voltammograms (CV’s) of a GC/Ni electrode during 50 cycles in 0.1 mol/L NaOH solution recorded at a potential sweep rate of 100 mV/s. In the first sweep, a pair of redox peaks appears at 480 and 390 mV/(vs Ag/AgCl); AgCl is assigned to Ni2+/ Ni3+redox couple in alkaline media. In the subsequent cycles, the peaks shift cathodically and stabilize at 451 and 378 mV/(vs Ag/AgCl), respectively. The entire behaviour is in accord once with the data reported previously in the literature concerning the formation and interconversion of α- and β-phase of Ni(OH)2, its conversion to NiOOH and the enrichment of Ni3+species on or just beneath the surface [18, 19, 26-28].

Fig. 3 SEM images of bare GC (a), GC/Ni (b), and GC/NiCu (c)
Figure 5 shows the consecutive CV’s of the GC/NiCu electrode in 0.1 mol/L NaOH solution recorded at a potential sweep rate of 100 mV/s. The oxygen evolution reaction is markedly impeded and its intensity remains unchanged with further polarization. Ni sites are considered responsible for the oxygen evolution reaction [18, 19, 29]. The first positive potential scan reveals a characteristic similar to that of pure Ni. Initially,a Ni(OH)2 layer is formed on the surface of GC/NiCu alloy electrode during the positive potential scan [24]. The rapid formation of Ni(OH)2 at low potential leads to a Cu-rich metal surface, which is oxidized to Cu2O later and completely to Cu(OH)2 at longer time. Therefore, the surface layer is subsequently transformed into a mixture of NiOOH and Cu(OH)2. In addition, some Cu(OH)2 and CuO can be oxidized further to the Cu3+oxide prior to evaluation of oxygen [30]. The change observed in the current of oxygen evolution may refer in part to the overpotential for the oxygen evolution reaction on copper oxides [24, 30]. In consecutive CV’s of GC/NiCu alloy electrode, the peak potentials, except the first cycle, are invariable, suggesting that the phase transformation of Ni oxyhydroxide from β to γ is inhibited. It is reported that there are four phases produced over the lifetime of a nickel hydroxide electrode substrate, namely, β-Ni(OH)2, α-Ni(OH)2, β-NiOOH, and γ-NiOOH [18, 19, 24].
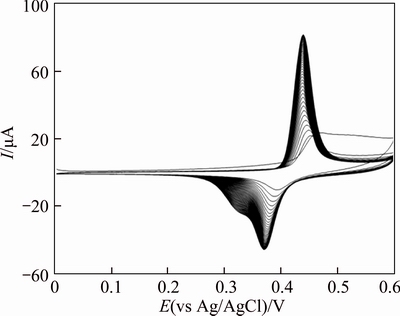
Fig. 4 Consecutive CV’s (50 cycles) of GC/Ni in 0.1 mol/L NaOH at a scan rate of 100 mV/s
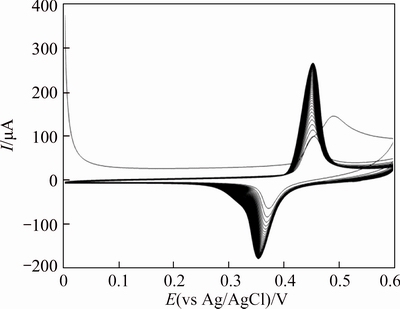
Fig. 5 Consecutive CV’s (50 cycles) of GC/NiCu electrode in 0.1 mol/L NaOH at a scan rate of 100 mV/s
The well-known Bode diagram identifies the phase transformations that are likely to occur during a normal cycle. β-Ni(OH)2 is first oxidized into β-NiOOH and the subsequent reduction during the cell discharge can be considered the intercalation into the b phase of one proton and one electron per Ni atom. Alternatively, γ-NiOOH is reduced into the hydrated α-Ni(OH)2 phase, which is unstable in strong alkali and ages to the β-form. In addition, β-NiOOH is partially converted to γ-NiOOH under experimental conditions of overcharge, high charge/discharge rates and high alkali concentration [31, 32]. However, these physical transformations occur slowly and are generally incomplete, so that α/γ and β/β systems coexist under steady state conditions. It is well-known that the formation of γ-NiOOH phase is associated with swelling or volume expansion of nickel film electrodes with subsequent micro cracks and disintegration of the nickel film. Lower inter electrode spacing results in lower internal resistance and therefore better efficiency of the electrode. Therefore, β-NiOOH phase is expected to be a better electroactive material for high electrochemical performance in alkaline solution. The comparison of the electrochemical behaviour of the pure electrodeposited Ni and Ni–Cu electrodeposited alloy (see Figs. 3 and 4, respectively) reveals the lower α/γ redox contribution with a good stabilization of β/β nickel oxyhydroxide form on the GC/Ni electrode. This is due to inhibition of peak potential variation after prolonging electrochemical treatment in alkaline solution. Thus, the addition of copper hydroxide to the nickel oxyhydroxide species represents a very efficient strategy of suppressing formation of γ-NiOOH phase. Similar electrochemical behaviour was observed for alloys obtained by the addition of several percent of Cd, Zn, Co, Ca, Ti, and other elements as solid solutions to the nickel hydroxide [32, 33].
Figures 6(a) and 7(a) present typical CV’s of a GC/Ni and GC/NiCu electrodes in 0.1 mol/L NaOH solution at various potential sweep rates of 2-2000 mV/s.
The peak currents are proportional to sweep rates in the range of 2-100 mV/s, Figures 6(b), (c) and 7(b) pointing to the electrochemical activity of the surface redox couple. From the slope of these lines and using [34]:
 (1)
(1)
where Г* is the surface coverage of the redox species and v is the potential sweep rate taking an average of both cathodic and anodic results; Г* values of around 1.6×10-8 and 3.1×10-8 mol/cm2 for GC/Ni and GC/NiCu electrodes respectively have been derived. These values correspond to the presence of around 40 and 65 monolayers of surface species for Ni and NiCu, respectively. In the higher potential sweep rates, this dependency is of the square root form. Figures 6(d), (e) signify the dominance of the diffusion controlled processes.
Figure 8 shows CV’s of GC/Ni and GC/NiCu electrode in 0.1 mol/L NaOH solution in the absence and presence of 1 mmol/L MFA at a potential sweep rate of 10 mV/s. As can be seen in 1 mmol/L MFA, GC/NiCu electrode generates higher current density for electro- oxidation of MFA around 500 mV in NaOH solution. In the presence of the MFA, it was observed that the anodic current and the associated anodic charge increased drastically, while the cathodic current and the corresponding charge decreased. In the presence of MFA, the anodic charge associated with the anodic peak is quantitatively 88.46% that of the corresponding cathodic peak, while in the absence of the MFA, it is 14.28%. This result indicates that MFA is oxidized by active nickel moiety through a cyclic mediation redox process. Nickel species are immobilized on the electrode surface, and the one with a higher valence oxidizes MFA via a chemical reaction followed by generation of low-valence nickel. Along this line, the high-valence oxide is regenerated through the external electrical circuit. Accordingly, MFA is oxidized via an EC' mechanism [34]. Moreover, the significant current in the reverse sweep indicates that the reaction of MFA with high- valence nickel oxide is the rate-determining step of the oxidation process. In Fig. 8, curves 2 and 3, also indicate that the onset potential of oxidation of low-valence nickel oxide decreases in the presence of the MFA, suggesting the facilitation of nickel oxidation of low valence oxide by MFA. The larger MFA response of the GC/NiCu electrode in comparison with GC/Ni electrode is proposed to be the result of pre-adsorption of MFA molecules at Cu sites by interaction of non-bonded electron pairs on the O-atom of MFA with the partially vacant d-orbital of the Cu species within the oxide film on the pre-anodized alloy surface and high amount of β-NiOOH on the surface of GC/NiCu in comparison with GC/Ni electrode.
Figure 9(a) shows cyclic voltammograms of GC/NiCu electrode in 0.1 mol/L NaOH solution in the presence of various concentrations of MFA at a potential sweep rate of 10 mV/s. At GC/NiCu electrode, oxidation of MFA appeared as a typical electrocatalytic response. The anodic charge increased with respect to that observed in the modified surface in the absence of MFA and it was followed by decreasing the cathodic charge upon increasing the concentration of the drug in solution.The anodic current in the positive sweep was proportional to the bulk concentration of MFA (Fig. 9(b)) and any increase in the concentration of MFA caused an almost proportional linear enhancement of the anodic current. So, catalytic electrooxidation of the MFA on GC/NiCu seems to be certain. The anodic peak current depending on concentration plot suggests a linear behaviour in typical calibration range of 0.25-1.5 mmol/L with a correlation coefficient of 0.993. The R.S.D. values ranging from 0.42% to 9.24% were obtained for three calibration curves. Ipa=1.148c+ 0.5847(R2=0.993) was used as a typical regression equation for the quantization. Method precision was evaluated regarding the both reproducibility (multiple electrodes) repeatability (single electrode). Repeatability was examined by continuous electrocatalytic MFA oxidation using same GC/NiCu electrode for 6 analyses in a standard solution. The anodic peak current was reduced by 1.2% after 6 complete scans which proved that the electrode has good repeatability and thereupon, surface poisoning did not occur. Reproducibility was determined by analysis of three replicate standard samples containing 0.25, 0.75 and 1.25 mmol/L of MFA in 3 consecutive days. In this regard, the percent R.S.D. values ranging from 2.45% to 8.06% were obtained.
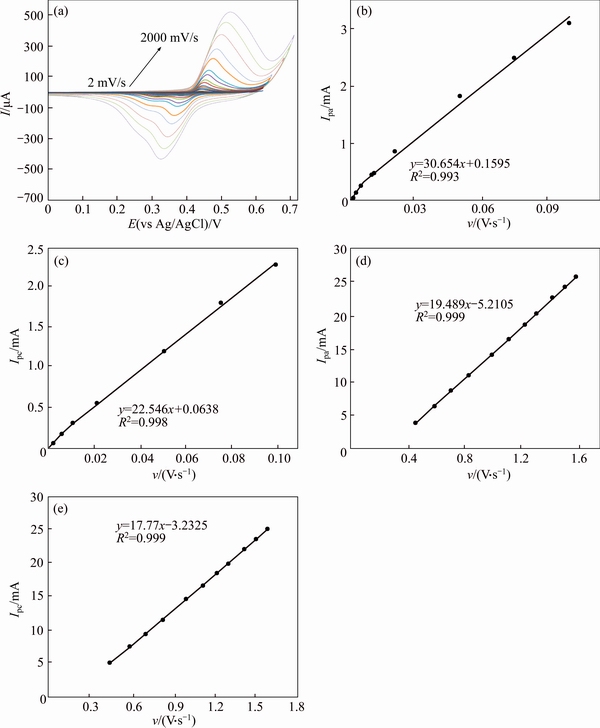
Fig. 6 Typical CV’s of a GC/Ni electrode in 0.1 mol/L NaOH in the potential sweep rates of 2, 5, 7, 10, 20, 30, 40, 50, 75, 100, 200, 350, 500, 750, 1000, 1250, 1500, 1750, 2000 mV/s (a), dependency of anodic (b) and cathodic peak currents to the sweep rate at lower values (2-100 mV/s) (c) and proportionality of anodic (d) and cathodic peak currents to the square roots of sweep rate at higher values (50-2000 mV/s) (e)
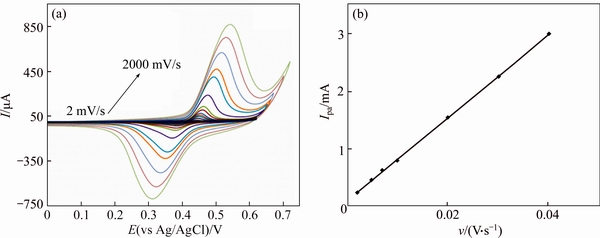
Fig. 7 Typical CV’s of a GC/NiCu electrode in 0.1 mol/L NaOH in potential sweep rates of 2, 5, 7, 10, 20, 30, 40, 50, 75, 100, 200, 350, 500, 750, 1000, 1250, 1500, 1750, 2000 mV/s (a) and dependency of anodic peak currents to sweep rate at lower values (2-40 mV/ s) (b)
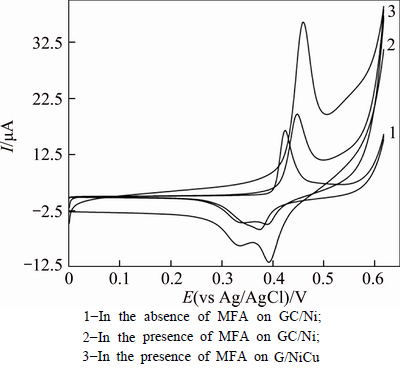
Fig. 8 Cyclic voltammograms in the absence and presence of 1 mmol/L of MFA on GC/Ni and GC/NiCu electrode in 0.1 mol/L NaOH solution (The potential sweep rate was 10 mV/s):
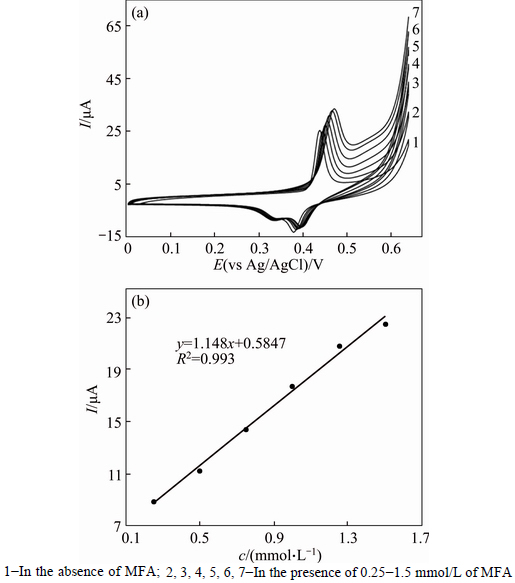
Fig. 9 Cyclic voltammograms of GC/NiCu electrode in 0.1 mol/L NaOH solution in the absence and presence of 0.25-1.5 mmol/L of MFA in solution (The potential sweep rate was 10 mV/s) (a), and dependency of anodic peak current on concentration of MFA solution (b):
Figure 10 presents cyclic voltammograms for 0.5 mmol/L MFA recorded using a GC/NiCu electrode at different potential sweep rates. The anodic peak currents increased linearly with the square root of the corresponding potential sweep rate (Fig. 10(a)), which indicates a mass transfer-controlling process of oxidation via diffusion.
In addition, the value of the electron-transfer coefficient (α) for the reaction can be obtained from the equation [34]:
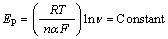 (2)
(2)
which is valid for a totally irreversible diffusion- controlled process. Using the dependency of anodic peak potential on the natural logarithm of the potential sweep rate (Fig. 10(c)), we obtained 0.68 and 0.85 as the values of the electron-transfer coefficients for MFA in GC/Ni and GC/NiCu electrodes respectively. On the basis of the slopes of the linear dependence of the anodic peak currents on the square root of the potential sweep rates (Fig. 10(b)), and the Randles–Sevcik equation [34]:
 (3)
(3)
where Ip is the peak current; A is the electrode surface area; D is the diffusion coefficient and C is the bulk concentration of MFA; the diffusion coefficients for MFA were calculated to be 8.3×10-6 and 9.2×10-6 cm2/s, for GC/Ni and GC/NiCu electrodes respectively, which agree with the literature values [24, 35].
Chronoamperograms were recorded by setting the working electrode potentials to the desired values and measuring the catalytic rate constant on the GC/Ni and GC/NiCu electrodes surface. Figure 11 depicts chronoamperograms for the GC/NiCu electrode in the absence (curve 1) and presence (curves 2-6) of MFA over the concentration range of 0.25-1.50 mmol/L. The applied potential steps were 320 mV and 560 mV, respectively. Plotting the net currents versus the minus square roots of time results in linear dependencies(Fig. 11, inset). Therefore, a diffusion-controlled process is dominant for electrooxidation of the MFA, as demonstrated previously using CV. By using the slopes of these lines, we can obtain the diffusion coefficients of the MFA according to the Cottrell equation [34]:
I=nFAD1/2Cπ-1/2t-1/2 (4)
where D is the diffusion coefficient; C is the bulk concentration. The mean values of the diffusion coefficients for MFA were 8.45×10-6 and 9.15×10-6 cm2/s on the GC/Ni and GC/NiCu surface, respectively. These values are in agreement with those obtained using CV (Fig. 10).
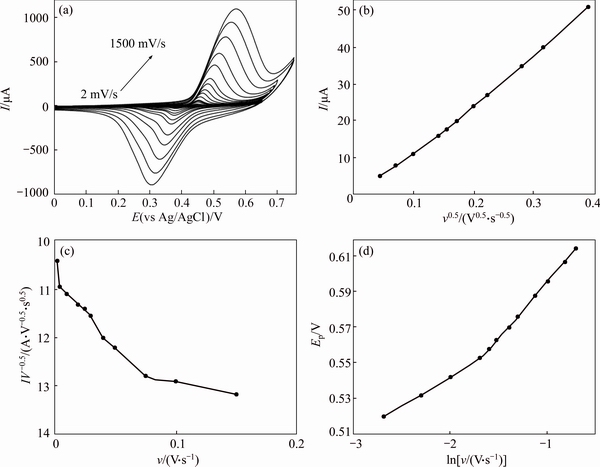
Fig. 10 Typical cyclic voltammograms of GC/NiCu electrode in 0.1 mol/L NaOH in the presence of 0.5 mmol/L MFA at various potential sweep rates of 2, 5, 7, 10, 20, 30, 40, 50, 75, 100, 200, 350, 500, 750, 1000, 1250 and 1500 mV/s (a), dependence of anodic peak current during the forward sweep on square roots of sweep rate (b), anodic current function (I/ν1/2) vs potential sweep rate v (c) and dependence of peak potential on lnν for oxidation of MFA at GC/NiCu electrode (d)
Chronoamperometry can also be used to evaluate the catalytic rate constant according to [34]:
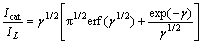 (5)
(5)
where Icat and IL are the currents in the presence and absence of the drug, respectively; γ=k0Ct is the argument of the error function; k0 is the catalytic rate constant; t is elapsed time. In cases where γ>1.5, erf(γ1/2) is almost equal to unity, and Eq. (5) can be reduced to [34]
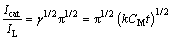 (6)
(6)
From the slopes of the Icat/IL versus t1/2 plots in Figs. 11, 12, the mean values of k0 obtained for MFA on GC/NiCu and GC/Ni surface were 5.4×105 and 5.8×105 cm3/(mol·s), respectively.
Based on the reported results and presented mechanism concerning the electrocatalytic activity of Ni in Refs. [18-24], the following mechanism can be proposed for the mediated oxidation of the drugs on the GC/NiCu electrode surface. The corresponding kinetics is also formulated. The redox transition of the nickel species,
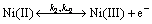 (7)
(7)
is followed by the oxidation of drugs on the modified surface via the reaction:
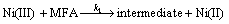 (8)
(8)
 (9)
(9)
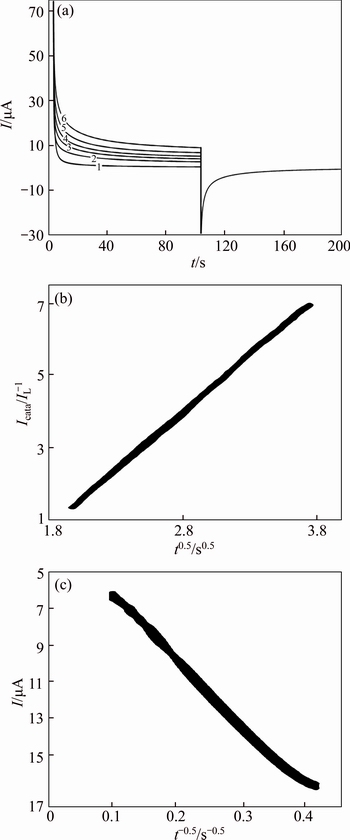
Fig. 11 Double steps chronoamperograms of GC/Ni electrode in 0.1 mol/L NaOH solution with different concentrations of MFA (1-0; 2-0.25 mmol/L; 3-0.5 mmol/L; 4-0.75 mmol/L; 5-1.0 mmol/L; 6-1.5 mmol/L). Potential steps were 320 mV and 560 mV, respectively (a) and dependency of transient current on t1/2 (b) and t -1/2 (c)
Typical amperometric signals obtained during successive increments of MFA are depicted in Fig. 13. Gentle stirring for a few seconds was needed to promote solution homogenization after each injection. The electrode response is quite rapid and proportional to the MFA concentration. The limits of detection (LOD) of the procedure were calculated according to the 3S.D./m criteria, where S.D. is the standard deviation of the intercept and m is the slope of the calibration curves [36]. The value of (3.13±0.13)×10-5 mol/L was determined for MFA LOD on the surface of the GC/NiCu electrode.
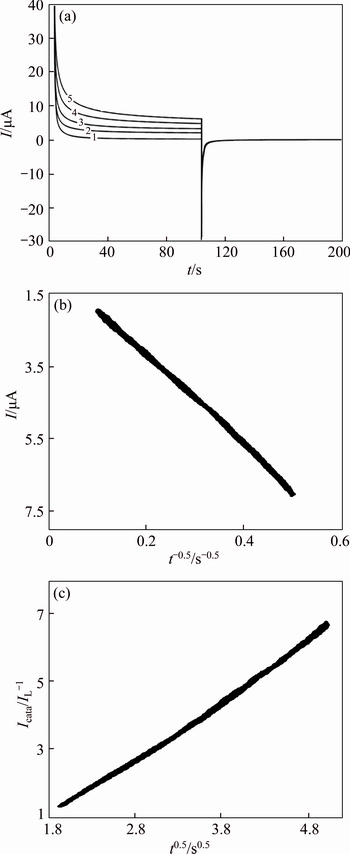
Fig. 12 Double steps chronoamperograms of GC/NiCu electrode in 0.1 mol/L NaOH solution with different concentrations of MFA of different concentrations of MFA (1-0 mol/L; 2-0.25 mmol/L, 3-0.5 mmol/L, 4-0.75 mmol/L and 5-1.0 mmol/L. Potential steps were 320 mV and 560 mV, respectively) (a) and dependency of transient current on t -1/2 (b) and t1/2 (c)
With the intention of confirming the application of the proposed method, the MFA amount determination in commercial capsules was performed. The development of voltammetric procedures for the electrocatalytic oxidation of MFA by modified electrodes was in the center of attention in previous years because of their importance in quality control of drug products. However,in previous years, because of the significance of MFA determination in quality control of drug products, obtaining a voltammetric procedure for the electrocatalytic oxidation of MFA using modified electrodes was highly considered. According to our knowledge, no document, using this GC/NiCu electrode, has been reported yet. The result of the assay of MFA capsules yielded a recovery of 96.8% (R.S.D.=4.9%) of label claim with a percent relevant error of –3.2%.
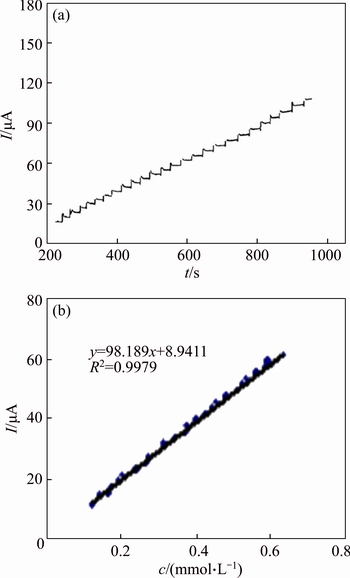
Fig. 13 Main panel: Current signal as a function of time in 0.1 mol/L NaOH solution during repetitive injections of MFA. E=500 mV (a) and dependency of peak current on MFA concentration (b)
4 Conclusions
Ni and NiCu alloy modified glassy carbon electrodes were checked for electrooxidation of the MFA in alkaline medium. These electrodes prepared by galvanostatic and different electrochemical techniques such as cyclic voltammetry, chronoamperometry were used to track the oxidation process and its kinetics. The electrode showed electrocatalytic oxidation of MFA. Chronoamperometric studies demonstrated a large anodic current at the oxidation potential of low-valence nickel hydroxide, in further support of the mediated electrooxidation. Using cyclic voltammetry and chronoamperometry techniques, we determined the kinetic parameters of these drugs, such as charge-transfer coefficient, catalytic reaction rate constant, and diffusion coefficient for oxidation. Sufficient experimental results demonstrated that the electrode can be applied in routine analysis of MFA and as a detector for detecting systems. Therefore, the electrode has the potential to be used for the successful determination of the MFA in pharmaceutical preparations.
Acknowledgements
The authors would like to acknowledge financial assistance from Tehran University of Medical Sciences, Tehran, Iran.
References
[1] REMINGTON J P, TROY D B, BERINGER P. Remington: The science and practice of pharmacy [M]. New York: Lippincott Williams & Wilkins, 2006.
[2] BRAYFIELD A. Martindale: The complete drug reference [M]. New York: Pharmaceutical Press, 2014.
[3] SHAH D A, RANA J P, BALDANIA S L, CHHALDTIYA U K, BHATT K K. High-performance thin-layer chromatographic method for the estimation of paracetamol, dicyclomine hydrochloride, and mefenamic acid in combined tablet dosage form [J]. JPC-Journal of Planar Chromatography-Modern TLC, 2014, 27: 52-57.
[4] AL-ABACHI M Q, HADI H. Simple, rapid and sensitive method for the determination of mefenamic acid in pharmaceutical preparations [J]. Journal of Analytical Chemistry, 2014, 69: 769-776.
[5] BEIRAGHI A, POURGHAZI K, AMOLI-DIVA M, RAZMARA A. Magnetic solid phase extraction of mefenamic acid from biological samples based on the formation of mixed hemimicelle aggregates on Fe3O4 nanoparticles prior to its HPLC-UV detection [J]. Journal of Chromatography B, 2014, 945: 46-52.
[6] ZISIMOPOULOS E G, TSOGAS G Z, GIOKAS D L, KAPAKOGLOU N I, VLESSIDIS A G. Indirect chemiluminescence-based detection of mefenamic acid in pharmaceutical formulations by flow injection analysis and effect of gold nanocatalysts [J]. Talanta, 2009, 79: 893-899.
[7] PEREZ-RUIZ T, MARTINZ-LOZANO C, SANZ A, BRAVO E. Determination of flufenamic, meclofenamic and mefenamic acids by capillary electrophoresis using β-cyclodextrin [J]. Journal of Chromatography B: Biomedical Sciences and Applications, 1998, 708: 249-256.
[8] MOREIRA A P L, MARTINI M, CARVALHO L M. Capillary electrophoretic methods for the screening and determination of pharmacologic adulterants in herbal-based pharmaceutical formulations [J]. Electrophoresis, 2014, 35: 3212-3230.
[9] SONG J F, GUO W, KANG X F, HU Y H. Investigation and application of polarographic catalytic wave of oxygen reduction caused by mefenamic acid [J]. Sci China B, 1993, 36: 906-911.
[10] BLANCO-LOPEZ M C, LOBO-CASTANON M J, MIRANDA- ORDIERES A J,  P. Voltammetric response of diclofenac-molecularly imprinted film modified carbon electrodes [J]. Anal Bioanal Chem, 2003, 377: 257-261.
P. Voltammetric response of diclofenac-molecularly imprinted film modified carbon electrodes [J]. Anal Bioanal Chem, 2003, 377: 257-261.
[11] AHMADI M, MADRAKIAN T, AFKHAMI A. Molecularly imprinted polymer coated magnetite nanoparticles as an efficient mefenamic acid resonance light scattering nanosensor [J]. Anal Chim Acta, 2014, 852: 250-256.
[12] MOGHADDAM A B, MOHAMMADI A, MOHAMMADI S. Electroanalysis of mefenamic acid in pharmaceutical formulation and spiked biological fluids on modified carbon nanotube electrode [J]. Pharmaceut Anal Acta, 2012, 3: 6-10.
[13] RIYANTO A, ANSHORI A. Electroanalysis of mefenamic acid using platinum powder composite microelectrode (PPCM) [J]. Anal Bioanal Chem, 2014, 6: 159-169.
[14] DOU Z, CUI L, HE X. Electrochimical determination of uric acid, xanthine and hypoxanthine by poly(xylitol) modified glassy carbon electrode [J]. J Cent South Univ T, 2014, 21(3): 870-876.
[15] MOHAMMADI A, MOGHADDAM A B, ALIKHANI E, EILKHANIZHDEH K, MOZAFFARI S. Electrochemical quantification of fluoxetine in pharmaceutical formulation using carbon nanoparticles [J]. Micro Nano Lett, IET, 2013, 8: 853-857.
[16] MOGHADDAM A B, MOHAMMADI A, MOHAMMADI S, RAYEJI D, DINARVAND R, BAGAI M, WALKER R B. The determination of acetaminophen using a carbon nanotube: Graphite-based electrode [J]. Microchim Acta, 2010, 171: 377-384.
[17] GHORBANI-BIDKORBEH F, SHAHROKHIAN S, MOHAMMADI A, DINARVAND R. Simultaneous voltammetric determination of tramadol and acetaminophen using carbon nanoparticles modified glassy carbon electrode [J]. Electrochim. Acta, 2010, 55: 2752-2759.
[18] SHAHROKHIAN S, GHORBANI-BIDKORBEH F, MOHAMMADI A, DINARVAND R. Electrochemical determinations of 6-mercaptopurine on the surface of a carbon nanotube-paste electrode modified with a cobalt salophen complex [J]. J Solid State Electrochem, 2012, 16: 1643-1650.
[19] NAEEMY A, MOHAMMADI A, BAKHTIARI H, ASHOURI N, WALKER R B. Electro- oxidation of acetaminophen on nickel/poly (O-aminophenol)/ multi-walled carbon nanotube nanocomposite modified graphite electrode [J]. Micro Nano Lett, IET, 2014, 9: 691-696.
[20] MOHAMMADI A, MOGHADDAM A B, BADRAGHI J. Direct electron transfer of ferritin on electrodeposited nickel oxide cubic nanoparticles [J]. Anal Methods, 2012, 4: 1024-1028.
[21] MOHAMMADI A, MOGHADDAM A B, KAZEMZAD M, DINARVAND R, BADRAGHI J. Synthesis of nickel oxides nanoparticles on glassy carbon as an electron transfer facilitator for horseradish peroxidase: Direct electron transfer and H2O2 determination [J]. Mater Sci Eng C, 2009, 29: 1752-1758.
[22] EHSANI A, MAHJANI M G, JAFARIAN M, NAEEMY A. Influence of ionic surfactant on physio-electrochemical properties and fractal dimension of poly ortho aminophenol film [J]. Prog Org Coat., 2010, 69: 510-516.
[23] EHSANI A, MAHJANI M G, JAFARIAN M, NAEEMY A. Electrosynthesis of polypyrrole composite film and electrocatalytic oxidation of ethanol [J]. Electrochim Acta, 2012, 71: 128-133.
[24] FEIZBAKHSH A, EHSANI A, NAEEMY A. Electrocatalytic oxidation of paracetamol on Ni and NiCu alloy modified glassy carbon electrode [J]. J Chinese Chem Soc, 2012, 59: 1086-1093.
[25] KHULBE K, MANN R, MANOOGIAN A. Behavior of nickel-copper alloy in hydrogenation, orthohydrogen-parahydrogen conversion and H2-D2exchange reaction [J]. Chem Rev, 1980, 80: 417-428.
[26] BRIGGS G, SNODIN P. Ageing and the diffusion process at the nickel hydroxide electrode [J]. Electrochim Acta, 1982, 27: 565-572.
[27] HAHN F, BEDEN B, CROISSANT M, LAMY C. In situ UV visible reflectance spectroscopic investigation of the nickel electrode- alkaline solution interface [J]. Electrochim Acta, 1986, 31: 335-342.
[28] DESILVESTRO J, CORRIGAN D A, WEAVER M J. Characterization of redox states of nickel hydroxide film electrodes by in situ surface Raman spectroscopy [J].J Electrochem Soc, 1988, 135: 885-892.
[29] CHEN S, BROWN L, LEVENDORF M, CAI W JU S Y, EDGEWORTH J, LI X, MAGNUSON C W, VELAMAKANNI A, PINER R D. Oxidation resistance of graphene-coated Cu and Cu/Ni alloy [J]. ACS Nano, 2011, 5: 1321-1327.
[30] LUO P, PRABHU S V, BALDWIN R P. Constant potential amperometric detection at a copper-based electrode: Electrode formation and operation [J]. Anal Chem, 1990, 62: 752-755.
[31] CHEN J, BRADHURST D, DOU S. Nickel hydroxide as an active material for the positive electrode in rechargeable alkaline batteries [J]. J Electrochem Soc, 1999, 146: 3606-3612.
[32] SINGH D. Characteristics and Effects of γ-NiOOH on cell performance and a method to quantify it in nickel electrodes [J]. J Electrochem Soc, 1998, 145: 116-120.
[33] LUO P F, KUWANA T, PAUL D K. Electrochemical and XPS study of the nickel-titanium electrode surface [J]. Anal Chem, 1996, 68: 3330-3337.
[34] BARD A J, FAULKNER L R. Electrochemical methods: Fundamentals and applications [M]. Vol. 2. New York: Wiley, 1980.
[35] HELI H, JABBARI A, MAJDI S, MAHJOUB M, MOOSAVI- MOVAHEDI A, SHEIBANI S. Electrooxidation and determination of some non-steroidal anti-inflammatory drugs on nanoparticles of Ni–curcumin-complex-modified electrode [J]. J Solid State Electrochem, 2009, 13: 1951-1958.
[36] MILLER J C, MILLER J N. Statistics for analytical chemistry [M]. 4th ed. New York: Ellis-Harwood, 1994.
(Edited by YANG Hua)
Cite this article as: Naeemy Ali, Mohammadi Ali, Ehsani Ali, Aghassi Ali. Electrocatalytic behaviour of Ni and NiCu alloy modified glassy carbon electrode in electro-oxidation of Contraflam [J]. Journal of Central South University, 2017, 24(8): 1703-1712. DOI: https://doi.org/10.1007/s11771-017-3577-7.
Received date: 2016-03-18; Accepted date: 2016-09-23
Corresponding author: Mohammadi Ali, PhD; Tel: +98-21-88358801; E-mail: Alimohammadi@tums.ac.ir

