MoSi2和WSi2电子结构及光学性质的数值研究
刘小良1, 2,赵中伟1,徐 慧2,李燕峰2
(1. 中南大学 冶金科学与工程学院,湖南 长沙,410083;
2. 中南大学 物理科学与技术学院,湖南 长沙,410083)
摘 要:应用基于密度泛函平面波赝势方法(PWP),考虑广义梯度近似(GGA)下的交换关联势,计算具有C11b型体心立方结构的MoSi2和WSi2单晶的电子态密度、能带结构、介电函数、吸收系数和折射率等电子结构及光学特性参量。计算结果表明:该类晶体的价带和导带部分重合,具有典型半金属特性,其费米面附近的态密度主要是由Mo或W原子中的d电子和Si原子3p态杂化而成,对晶体导电性贡献最大的是Mo和W原子中的d电子,其光学性质表现出各向异性,沿c轴方向介电函数和折射率都存在1个向低能方向偏移(红移)且峰值较大的峰;具有C11b型结构的MoSi2和WSi2由于Mo和W原子价电子不同导致其电子结构和光学性质存在微小差别。
关键词:平面波赝势方法;电子结构;光学性质
中图分类号:O472;O73 文献标识码:A 文章编号:1672-7207(2009)03-0568-07
Numerical investigation on electronic structures and optical properties of MoSi2 and WSi2 single crystals
LIU Xiao-liang1, 2, ZHAO Zhong-wei1, XU Hui2, LI Yan-feng2
(1. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China;
2. School of Physical Science and Technology, Central South University, Changsha 410083, China)
Abstract: The electronic structures and optical properties of MoSi2 and WSi2 single crystals with the C11b structure were computed by using the plane pseudo-potential method based on the density functional theory. The generalized-gradient approximate (GGA) was used for the exchange-correlation potential. The calculation result shows that those MoSi2 and WSi2 single crystals are of characteristics of a half-metal owing to the partial overlaps of the valence and conduction-band and it is d electrons in atom Mo or W that play a main role in the electrical conductivity. For the optical properties, the anisotropic of these models is proved by simulation results and the dielectric function, the absorption spectra and the refractive index present different behaviors for polarized light in different directions. In addition, while having the similar properties, the electronic structures and optical properties have subtle differences between MoSi2 and WSi2 because of the difference in the structure.
Key words: plane pseudo-potential method; electronic structure; optical property
MoSi2和WSi2是Mo(或W)和Si二元合金系中含硅量最高的一种中间相,具有金属和陶瓷的双重特性,融合了作为高温材料的许多优良属性如高熔点,极好的高温抗氧化性,良好的导热和导电性以及较低的线膨胀系数等。基于MoSi2和WSi2的材料是集功能和结构于一体的材料,被认为是继镍钼超合金和结构陶瓷之后出现的极具竞争力的高温结构材料[1-2]。20世纪60年代瑞典的康泰尔公司研制出可在1 773 K以上空气中使用的硅钼棒高温发热元件,目前,该材料已经实现了工业化生产和应用,成为高温发热元件的主 流[3-4],并已在航空航天、工业、电热材料及元件等诸多领域显示了其广阔的应用前景。在航空航天领域,可作为涡轮发动机的耐热部件,如叶片、燃烧室、喷嘴等;在工业领域的应用包括用于制造高温热交换器、气体燃烧器、火花塞、高温滤网以及作为制造熔炉的结构材料等。
从理论上对MoSi2和WSi2进行深入系统化研究存在一定困难,目前人们对MoSi2和WSi2的研究主要集中在实验研究方面[5-7]。随着第一性原理方法的发展、完善及其在其他材料上的应用成功[8-11],激发了人们对MoSi2和WSi2各种性质的理论研究,但是,由于MoSi2和WSi2在实践中主要是用作结构材料及发热元件,目前,更多的研究主要集中在其热学[12]和力学性质[13]方面,而对其电子结构和光学性质的理论研究很少。为此,本文作者利用第一性原理方法,针对具有C11b型体心立方结构的MoSi2和WSi2晶体,从理论上研究其电子结构和光学性质。
1 计算模型和方法
MoSi2和WSi2是一种道尔顿型金属间化合物,晶体通常呈四方C11b型体心立方结构[14],空间群为 ,群代号为139,在其晶体结构中,Mo―Mo(或W―W)之间是以金属键结合,Si―Si之间是以共价键结合,而Mo(或W)―Si之间既有金属键成分,又有共价键成分。MoSi2晶格常数的实验值[15]为a=b=0.320 2 nm,c=0.784 3 nm,而对于WSi2,a=b=0.321 8 nm,c=0.789 6 nm。
,群代号为139,在其晶体结构中,Mo―Mo(或W―W)之间是以金属键结合,Si―Si之间是以共价键结合,而Mo(或W)―Si之间既有金属键成分,又有共价键成分。MoSi2晶格常数的实验值[15]为a=b=0.320 2 nm,c=0.784 3 nm,而对于WSi2,a=b=0.321 8 nm,c=0.789 6 nm。
利用Material Studio软件中的CASTEP模块,并结合ABINIT计算软件,基于密度泛函的平面波赝势方法(PWP)[16-17],首先对晶格结构进行优化,并计算得到晶体的能带结构、电子态密度和跃迁矩阵元。对于其中的交换关联势,采用广义梯度近似(GGA)来描述,基函数借用Perdew等[18]的方案,平面波基矢的切断能量设置为290.0 eV,自洽循环计算的能量收敛标准为1.0×10-5 eV,K网格大小为8×8×4,不考虑温度效应。然后,从偶极跃迁矩阵元出发,进行晶体光学特性[19]研究,得到介电函数

最后,根据折射率和吸收系数与介电函数的相应关系可求出晶体的折射率和吸收系数。
2 电子结构
该计算是基于优化后的晶格参数所得,计算的第1步是利用CASTEP模块对晶格结构进行优化。优化后的晶格参数如下:对于MoSi2,a=b=0.319 0 nm,c= 0.779 5 nm;对于WSi2,a=b=0.321 0 nm,c=0.777 8 nm,对比实验值,优化后的晶格参数有所减小,但相对误差最大只有1.49%,在允许的误差范围之内。在优化的晶格参数基础上,为得到晶体的能带结构和电子态密度分布,可利用自洽迭代方法求解Kohn-Shame 方程:

多电子系统的能量可根据拉氏乘子直接计算得到。
图1所示为具有C11b型体心立方结构的MoSi2单晶的能带结构和电子态密度,图2所示为WSi2的对应结果。这里只显示了价电子和次外层电子的能带结构,图中虚线表示费米面的位置,费米面对应于能量的零点,其他各能带能量为相对于费米面的参照值。从图1和图2可看出,在费米面附近(见图1和图2中虚线位置),MoSi2和WSi2的价带和导带之间不存在明显的能隙,其价带和导带部分重叠,但重叠部分的态密度要明显小于其他导带和价带区域的态密度,因此,这2种化合物具有半金属的性质,其导电性能介于半导体和金属之间。

(a) MoSi2的能带结构;(b) MoSi2的电子态密度
图1 MoSi2的能带结构及电子态密度图
Fig.1 Energy bands and density of states of MoSi2

(a) WSi2的能带结构;(b) WSi2的电子态密度
图2 WSi2的能带结构及电子态密度图
Fig.2 Energy bands and density of states of WSi2
为突出费米面附近的电子结构特性,费米面附近MoSi2和WSi2的总电子态密度(DOS)和分态密度(PDOS)如图3所示。鉴于此例中s电子对费米面附近电子态密度贡献小,这里只画出了p电子和d电子的分态密度。由于Mo和W同族,它们与Si组成的化合物具有相似的结构,两者在费米面附近的态密度分布非常相似,在靠近费米面附近的低能成键态区域-5.0~1.0 eV和高能反键态区域1.0~7.5 eV各自存在1个态密度的尖峰。从图3还可以看出,存在1个区分低能成键态和高能反键态的区域-0.5~0.5 eV,这一区域内的态密度较小,约为0.4 态数/eV,表明该区域不存在带隙,价带和导带在这一区域部分交叠,印证了其半金属特性。2种晶体态密度曲线非常相似,几乎具有相同的价带宽度(约14 eV)和导带宽度(约15 eV)。但由于2种晶体中的Mo原子和W原子的价电子分别为4d55s1和5d46s2,导致其电子态密度存在一些细微的差异,体现在态密度峰值的形状及位置不同。从图3(a)和图3(b)中几个对应点M和M′,N和N′容易看出这两者的区别。

(a) MoSi2; (b) WSi2
图3 费米面附近MoSi2和WSi2的总电子态密度及p和d分态密度图
Fig.3 Total and partial densities of states of MoSi2 and WSi2 near Fermi surface
根据图3(a)中的各分态密度可以进一步看出,MoSi2在费米面附近的电子总态密度主要是由Mo原子的4d态和Si原子3p态所贡献,同样地,WSi2在
费米面附近的电子总态密度则主要是由W原子的5d态和Si原子3p态所贡献。通过对各原子轨道上电子布居数研究发现,在成键过程中,s轨道上的部分电子跃迁进入了p轨道和d轨道,进而参与了p态电子和d态电子的杂化作用。并且,相对来说,在靠近费米面区域,Mo(或W)原子的4d(或5d)态密度要大于Si原子的3p态密度,而在远离费米面区域,Si原子的3p态密度要大于Mo(或W)原子的4d(或5d)态密度,考虑到费米面附近电子对材料电学性质的决定性作用,因此,可以推测该类晶体的导电性能主要由d电子决定。
图4所示为2种化合物的总电子态密度与晶体各组成成分总电子态密度的对照情况,可以看出,Si原子在费米面附近的态密度很小,对化合物费米面附近态密度提供主要贡献是其中的Mo原子或W原子,准确地说,对化合物晶体导电性起主要作用的是Mo原子的4d电子或W原子的5d电子。

(a) MoSi2; (b) WSi2
图4 化合物MoSi2和WSi2的总电子态密度与各成分的电子态密度间的关系
Fig.4 Total densities of state for crystals of MoSi2, WSi2, Mo, W and Si
3 光学性质
3.1 介电函数
利用介电函数与跃迁矩阵之间的关系[19],可分别得到MoSi2和WSi2晶体介电函数的色散关系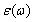 (图5)。计算中,剪刀算子值取0.5 eV,Lorentz展宽系数取为0.1 eV。容易看出,入射光偏振方向沿a轴、b轴方向,即垂直(100)和(010)面2个方向时,晶体具有相同的介电性质,但当入射光偏振方向沿c轴方向,即垂直(001)面方向时,晶体的介电函数与其他方向具有较大的差异。该类晶体介电函数的各向异性反映了其特殊的四方C11b型体心立方结构对称性,在该种结构中,沿a轴和b轴方向等价,但不等价于沿c轴方向。
(图5)。计算中,剪刀算子值取0.5 eV,Lorentz展宽系数取为0.1 eV。容易看出,入射光偏振方向沿a轴、b轴方向,即垂直(100)和(010)面2个方向时,晶体具有相同的介电性质,但当入射光偏振方向沿c轴方向,即垂直(001)面方向时,晶体的介电函数与其他方向具有较大的差异。该类晶体介电函数的各向异性反映了其特殊的四方C11b型体心立方结构对称性,在该种结构中,沿a轴和b轴方向等价,但不等价于沿c轴方向。

(a) MoSi2; (b) WSi2
图5 MoSi2和WSi2沿a轴(100),b轴(010)和c轴(001)方向的介电函数
Fig.5 Complex dielectric function for polarized light in crystals of MoSi2 and WSi2 along axes of a, b and c
就MoSi2介电函数的虚部 来说,当能量小于12 eV时,沿c轴方向的值总体大于沿a和b轴方向的值,沿c轴方向的介电函数的最大值达到60,而沿a和b轴方向的介电函数最大值只有40。沿a和b轴方向,介电函数在能量为4 eV和6 eV处有2个明显的峰值,但沿c轴方向只存在1个明显的峰,峰值位置明显前移到能量为3 eV处,且峰值较a和b轴方向有较大增加。就MoSi2的介电函数的实部
来说,当能量小于12 eV时,沿c轴方向的值总体大于沿a和b轴方向的值,沿c轴方向的介电函数的最大值达到60,而沿a和b轴方向的介电函数最大值只有40。沿a和b轴方向,介电函数在能量为4 eV和6 eV处有2个明显的峰值,但沿c轴方向只存在1个明显的峰,峰值位置明显前移到能量为3 eV处,且峰值较a和b轴方向有较大增加。就MoSi2的介电函数的实部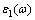 来说,沿a和b轴方向其正负转换的临界点为能量4 eV处,而沿c轴方向的临界点在能量3 eV处。对WSi2晶体,可得到相似的结论,从其介电函数曲线可以看出,沿a和b轴方向介电函数的虚部在能量区间4~6 eV具有较大的值,而在沿c轴方向介电函数的虚部的极值出现在能量约为3 eV附近,且峰值有较大增加。另外,WSi2晶体介电函数的实部正负转换的临界点与MoSi2的极为相近。
来说,沿a和b轴方向其正负转换的临界点为能量4 eV处,而沿c轴方向的临界点在能量3 eV处。对WSi2晶体,可得到相似的结论,从其介电函数曲线可以看出,沿a和b轴方向介电函数的虚部在能量区间4~6 eV具有较大的值,而在沿c轴方向介电函数的虚部的极值出现在能量约为3 eV附近,且峰值有较大增加。另外,WSi2晶体介电函数的实部正负转换的临界点与MoSi2的极为相近。
3.2 吸收系数
介电函数 与复折射率
与复折射率 之间存在关系为:
之间存在关系为:

部,习惯上称为折射率和消光系数,分别表征介质影响光传播时的相位特性和振幅特性,再由消光系数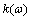 可得到晶体的吸收系数:
可得到晶体的吸收系数:

图6(a)所示为MoSi2对沿不同偏振光方向入射的吸收系数。对照图1中的能级结构,可以发现,图谱中处于高能区域的吸收谱对应于次外层电子能带向价电子能带之间的跃迁,而处于低能区域的吸收谱则对应于电子从价带向导带的跃迁。对应价带向导带跃迁的吸收谱的峰值出现在能量约为6.5 eV处,近似于图3中高能反键态密度峰值位置(E≈4.0 eV)和低能成键态密度峰值位置(E≈-2.5 eV)的能量差,说明吸收谱中的这一峰值正是由电子在这2个能级之间的跃迁形成的。精确的分析还可以看出吸收系数对方向的依赖性,沿c轴方向与沿a和b轴方向的吸收谱曲线存在着细微的差别,峰的高度和位置都存在一定的差异,特别在低频段,沿c轴方向在能量区间0~1.5 eV上的吸收系数很小,然后,吸收系数迅速增大,在能量为3 eV处形成1个吸收系数约为10的峰值(见图6(a)中的M处),考虑到可见光对应的能量范围为1.8~3.1 eV,可以推断MoSi2晶体在c轴方向对可见光的吸收要强于沿a轴和b轴方向的吸收。图6(b)所示为WSi2的吸收系数,从中容易得出和MoSi2相类似的吸收特性,另外,可以看出,WSi2和MoSi2的吸收谱曲线存在一定的差异,不仅表现在峰值的位置上,还体现在WSi2的吸收谱发生了一定程度的蓝移。
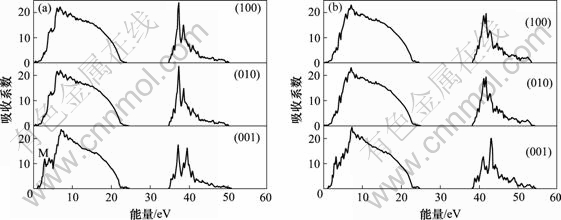
(a) MoSi2; (b) WSi2
图6 MoSi2和WSi2沿a轴(100),b轴(010)和c轴(001)方向的吸收系数
Fig.6 Polarized absorption spectra of MoSi2 and WSi2 along axes of a, b and c
3.3 折射率
由式(4)可得出晶体的折射率 的表达式为:
的表达式为:

图7(a)所示为MoSi2沿3个不同入射光偏振方向折射率的色散关系。可以看出,折射率在低能区趋向于5.0左右,在高能区趋向于1.0左右。在高能区域沿3个方向的折射率基本相同,但在低能区,沿c轴方向的折射率与沿a和b轴方向的折射率有较大差异。对于沿a和b轴方向,在能量区间0~4 eV上,折射率基本保持在5.0左右,超过4 eV以后才有1个快速下降阶段,但在c轴方向,当能量为2.5 eV时,折射率较大,约7.0,然后,随能量的增大折射率迅速减少。图7(b)所示为WSi2折射率的色散关系,与MoSi2的折射率曲线整体上相似,但也存在细微差别。

(a) MoSi2; (b) WSi2
图7 MoSi2和WSi2沿3个不同入射光偏振方向折射率的色散关系
Fig.7 Refractive index of MoSi2 and WSi2 along axes of a, b and c
4 结 论
a. 对具有C11b型体心立方结构的MoSi2和WSi2晶体总电子态密度、能带结构的模拟计算显示,该类晶体的价带和导带存在部分交叠,因而具有半金属特性,其导电性能介于半导体和金属之间。
b. 对晶体的导电性起主要贡献的是其中的Mo或者W原子,其导电性主要取决于Mo或者W原子中的d电子。
c. 该类晶体的光学性质表现出各向异性性,沿a轴和b轴方向等价,但不等价于沿c轴方向的光学 性质。
d. 具有C11b 型单晶结构的MoSi2和WSi2的电子结构和光学性质整体上相似,但它们的能带结构、态密度曲线、介电函数曲线以及吸收谱曲线存在一定的差别,说明其性质存在一定差别。
参考文献:
[1] Petrovic J J, Vasudevan A K. Key developments in high temperature structural silicides[J]. Mater Sci Eng A, 1999, A261(1/2): 1-5.
[2] Vasudevan A K, Petrovic J J. A comparative overview of molybdenum disilicide composites[J]. Materials Science and Engineering A, 1992, 155(1/2): 1-17.
[3] 江 莞, 赵世柯, 王 刚. 二硅化钼材料的研究现状及应用前景[J]. 无机材料学报, 2001, 16(4): 577-585.
JIANG Wan, ZHAO Shi-ke, WANG Gang. Progress in the research of molybdenum disilicide and its applications[J]. Journal of Inorganic Materials, 2001, 16(4): 577-585.
[4] 刘 芳, 范文捷, 陈玉超. 高品质硅钼电热元件的研制[J]. 耐火材料, 2003, 5(3): 304-305.
LIU Fang, FAN Wen-jie, CHEN Yu-chao. The preparation of high quality Si-Mo element in electro-heating[J]. Refractories, 2003, 5(3): 304-305.
[5] 郜剑英, 江 莞, 李建林, 等. 二硅化钼合金化及掺杂改性的研究进展[J]. 中国钼业, 2003, 27(4): 23-27.
GAO Jian-ying, JIANG Wan, Li Jian-lin, et al. Progress in alloying and doping of MoSi2[J]. China Molybdenum Industry, 2003, 27(4): 23-27.
[6] Petrovic J J. Toughening strategies for MoSi2-based high temperature structural silicides[J]. Intermetallics, 2000, 8(9/11): 1175-1182.
[7] Duman S, BaGci S, Tütüncü H M, et al. First-principles studies of ground-state and dynamics of MgS, MgSe and MgTe in the rocksalt, zinc blende, wurtzite, and nickel arsenide phase[J]. Physical Review B, 2006, 73(20): 205201.
[8] Segall M D, Lindan P J D, Probert M J, et al. First-principles simulation: ideas, illustrations and the CASTEP code[J]. J Phys: Cond Matt, 2002, 14(11): 2717-2743.
[9] 李燕峰, 徐 慧, 宋招权, 等. Zn掺杂对MgB2 电子结构及超导转变温度的影响[J]. 中南大学学报: 自然科学版, 2006, 37(5): 925-931.
LI Yan-feng, XU Hui, SONG Zhao-quan, et al. The effect of Zn doping on the electronic structure and superconductivity temperature of MgB2[J]. Journal of Central South University: Science and Technology, 2006, 37(5): 925-931.
[10] 刘廷禹, 张启仁, 庄松林. 含铅空位的PbWO4晶体光学性质及其偏振特性的研究[J]. 物理学报, 2005, 54(8): 3780-3786.
LIU Ting-yu, ZHANG Qi-ren, ZHUANG Song-lin. Optical polarized properties for the PbWO4 crystal containing lead vacancy[J]. Acta Physica Sinica, 2005, 54(8): 3780-3786.
[11] 张 宝, 郭华军, 李新海, 等. 量子化学从头计算法计算石墨微晶中碳原子的静电荷[J]. 中南大学学报: 自然科学版, 2006, 37(5): 919-924.
ZHANG Bao, GUO Hua-jun, LI Xin-hai, et al. Determination of net charge of carbon atoms in graphite crystallite by ab initio calculation method[J]. Journal of Central South University: Science and Technology, 2006, 37(5): 919-924.
[12] Kimura K, Nakamura M, Hirano T. High temperature deformation behavior of MoSi2 and WSi2 single crystals[J]. J Mater Sci, 1990, 25(5): 2487-2492.
[13] Nakamura M, Matsumoto S, Hirano T. Elastic constants of MoSi2 and WSi2 single crystals[J]. J Mater Sci, 1990, 25(7): 3309-3331.
[14] Umakoshi Y, Sakagami T, Hirano T, et al. High temperature deformation of of MoSi2 single crystals with the C11b structure[J]. Acta Metall Mater, 1990, 38(6): 909-915.
[15] 周 飞. MoSi2 和WSi2相结构和性能的电子理论研究[J]. 硅酸盐学报, 2000, 28(5): 462-464.
ZHOU Fei. Study on the structure and properties for MoSi2 and WSi2 phases by electron theory[J]. Journal of the Chinese Ceramic Society, 2000, 28(5): 462-464.
[16] Troullier N, Martins J L. Efficient pseudopotentials for plane-wave calculations[J]. Physical Review B, 1991, 43(3): 1993-2006.
[17] Kresse G, Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set[J]. Physical Review B, 1996, 54(16): 11169-11186.
[18] Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B, 1992, 45(23): 13244-13249.
[19] Ambros-Drachl C, Majewski J A, Vogl P, et al. First-principles studies of the structural and optical properties of crystalline poly (para-phenylene)[J]. Physical Review B, 1995, 51(15): 9668-9676.
[20] 方容川. 固体光谱学[M]. 合肥: 中国科学技术大学出版社, 2001.
FANG Rong-chuan. Spectroscopy of solid[M]. Hefei: China University of Science and Technology Press, 2001.
收稿日期:2008-08-03;修回日期:2008-12-01
基金项目:湖南省自然科学基金资助项目(08JJ3005);中国博士后科学基金资助项目(20080431025);中南大学博士后基金资助项目(2007年)
通信作者:赵中伟(1966-),男,教授,河北永年人,从事冶金物理化学及有色金属冶金研究;电话:0731-8830476;E-mail: zhongweizhao@hotmail.com

