���±�ţ�1004-0609(2014)11-2789-09
�����仯����L10-TiAl��ȱ��Ũ�ȵĵ�һԭ��
�ջԽ�1, 2����˳ƽ3��������1���� ͼ1���� ΰ1���� ��1, 2
(1. ���ϴ�ѧ ���Ͽ�ѧ�빤��ѧԺ����ɳ 410083��
2. ���ϴ�ѧ ��ɫ�������Ͽ�ѧ�빤�̽������ص�ʵ���ң���ɳ 410083��
3. ��������ѧԺ ���Ϲ���ѧԺ������ 213001)
ժ Ҫ�����õ�һԭ��ƽ�沨���Ʒ��������Wagner-Schottkyȱ������ѧģ�ͣ��о������仯����L10-TiAl�и��ֿ�λ�ͷ�λ��ȱ�ݵ��γ��ʡ�����ѧƽ��Ũ�ȼ�������õȡ������������Щȱ�ݵ�����ѧƽ��Ũ�Ⱦ����¶ȵ����߶��������з�λȱ��Ũ�Ⱦ����ڿ�λȱ��Ũ�ȣ�Ti��λŨ�ȸ���Al��λŨ�ȡ������뻯ѧ�����ȳɷ��£�Ti��λȱ�ݵ�Ũ����Al��λȱ�ݵĻ����൱������ƫ������ȵĸ�Ti�ɷֶˣ�Ti��λȱ�ݵ�Ũ�ȸ���Al��λȱ�ݵģ��ڸ�Al�ɷֶ����෴����ͬ��ȱ��֮����ձ������ų��ԣ����Ծۼ�����������������з�ɢ����ɢ��
�ؼ��ʣ�L10-TiAl�����仯�����ȱ��Ũ�ȣ��γ��ʣ���һԭ����Wagner-Schottkyģ��
��ͼ����ţ�TG111���� ���ױ�־�룺A
First principles of point defect concentrations in L10-TiAl intermetallic composite
TAO Hui-jin1, 2, SUN Shun-ping3, ZHANG Cheng-cheng1, CHEN Tu1, LUO Wei1, JIANG Yong1, 2
(1. School of Materials Science and Engineering, Central South University, Changsha 410083, China;
2. Key Laboratory of Nonferrous Metal Materials Science and Engineering,
Ministry of Education, Central South University, Changsha 410083, China;
3. School of Materials Engineering, Jiangsu University of Technology, Changzhou 213001, China)
Abstract: Using the plane wave pseudopotential method in first-principles and Wagner-Schottky model, the formation enthalpies, equilibrium concentrations, and interaction of vacancies and anti-site point defects were assessed for L10-TiAl intermetallics. The results show that, in the whole composition range of interest, all the defect concentrations increase with increasing temperature. In particular, the anti-site defect concentrations are higher than the vacancy defect concentrations, and the Ti vacancy concentration is higher than the Al vacancy concentration. At the stoichiometric composition, the Ti anti-site defect concentration is comparable to that of Al anti-site defect. At the Ti-rich side, the Ti anti-site defect concentration is higher than that of Al anti-site defect, while at the Al-rich side, the Al anti-site defect concentration is higher than that of Ti anti-site defect. The interaction between these defects is essentially repulsive, which facilitates the defect distribution and diffusion in the matrix.
Key words: L10-TiAl intermetallic composite; point defect concentration; formation enthalpy; first principles; Wagner-Schottky model�����仯��������ܶ�С��ǿ�ȸߺ�����ѧ���ܺõ��ŵ㣬��һ����Ҫ�ĺ��պ����ø��½ṹ����[1-2]���ر���TiAlϵ�����仯�����ѳɹ�Ӧ���ڷɻ����桢�����Լ�������ҡ�˵�������Ҫ�ṹ����[3-4]�������仯�����д��ڶ������͵�����ѧ��ȱ�ݣ������ʺ���Ϊ�Ƚϸ��ӣ���Ũ�����¶ȺͲ��ϳɷֵ�Ӱ��̶Ȳ�ͬ���Բ��ϵ��ȵ����絼����ѧ����ѧ����Ҫ�������Žϴ�Ӱ��[5-7]����������ʵ�鷽����չ��ȱ�ݺ͵�ȱ��Ũ�ȵ��о��������ѶȽϴɿ���Ҳ���ߡ������������ڵ�һԭ���IJ���ȱ�ݼ����о��������죬�ѳ�Ϊ�����仯�����ȱ�ݵ���Ҫ�о��ֶΣ���JIANG��[8-9]���ڵ�һԭ�������о���B2��NiAl��C15��NbCr2�еĵ�ȱ��Ũ����ռλ������Ni��λ��Ni��λ�ֱ��Ǹ�Al��Ni��B2-NiAl����ѧ���ȶ��ĵ�ȱ����ʽ�����ɽ���Zr��Hf��Ta�������ɷֺ��¶ȷ�Χ�ڶ�����ռ��C15��NbCr2��Nb�ǵ���Ti��V��Mo��W��ռλ��ȡ���ڳɷֺ��¶ȡ���˳ƽ��[10]������L12-Al3Li�����仯����ĵ�ȱ��Ũ�����¶Ⱥͳɷֵı仯������Al��Li��λȱ��Ũ�Ƚϸߣ�Li��λȱ��Ũ�Ƚϵͣ���Al��λŨ����С���¶����ߣ����ֵ�ȱ�ݵ�Ũ������KORZHAVYI��[11]�о�B2-NiAl������ѧ��ȱ������ã�����Ni��λ��Al��λ֮���Լ�Al��λ��Al��λ֮����ܴ���ǿ�ҵ����������Щ����ö�ȱ��ƽ��Ũ��Ӱ���С���������ڽ���B2-NiAl�е�����ȱ����̬��
��TiAlϵ�����仯���﷽�棬ZHU��[12]���ڵ�һԭ�������о�Al3Ti�е�ȱ���Լ�Siռλ�������ڸ�Al�ɷ��£�Al��λ��ȱ���γ�����ͣ�Si������ռ��Al��λ������Si��Al3Ti�д��������ܶȡ����ɵ�[13-14]�о�Ԥ��ͱȽ�3d���ɽ���Ԫ����L10-TiAl�е�ռλ������d���������ٵĹ���Ԫ��(Sc��V��Cr)����ռ�� Tiԭ���ǵ���, ��d�������϶���������d�Dz��Ԫ��������ռ�� Alԭ���ǵ���JIANG[15]������������Wagner-Schottkyͳ���� ��ѧģ�ͽ�һ���Ƚ�3d��4d��5d���ɽ���Ԫ����L10-TiAl��ѡ��ռλ������Zr��Hf����ռ��Tiԭ���ǵ���Co��Ru��Rh��Pd��Ag��Re��Os��Ir��Pt��Au����ռ��Alԭ���ǵ���V��Cr��Mn��Fe��Ni��Cu��Nb��Mo��Tc��Ta��Wԭ�ӵ�ռλ������ǿ�ҵ������ںϽ�ɷֵļ����Ⱥ��¶ȱ仯��LI��[16]���õ�һԭ��ƽ�沨���Ʒ����о���Nb L10-TiAl�Ͻ��е�ȱ��֮�������ã�����ռ��Alԭ���Ǹ���ϵ����������Nbԭ�Ӿ����ų����ã����ν���Nbԭ���������������Alԭ���Ǹ���ϵ�Nbԭ�ӿ����γɶ̳�����ṹ���⽫��������ߺ�NbTiAl�Ͻ��ǿ�ȡ�WANG��[17]���õ�һԭ������������Ti-Al��Ԫϵ������B2���в�ͬ��ȱ�ݵ��γ��ʣ��������Al�Ǹ�㱻Tiԭ��ռ���γɵķ�λȱ�ݺ�Ti�Ǹ�㱻��λռ���γɵĿ�λȱ���������ϸ������ƣ��ڴ˻����϶�B2�������ѧģ�ͽ��еĸĽ��������Ż��õ�����ʵ��ֵ���ϵø��õĽ����
��ǰ��L10-TiAl�е�����Ԫԭ�ӵ�ѡ����ռλ���н϶����Ԥ�⣬�����䱾����ȱ��(Ti��Al�Ŀ�λ���ַ�λ)Ũ�����¶ȺͳɷֵĹ�ϵ��������ȱ�ݼ������á��Լ���ȱ���γɼ����ܵȵ��о�ȴδ����������ˣ��������߽����õ�һԭ����������L10-TiAl�и�����Ҫ��ȱ�ݵ��γ��ʣ���һ�����Wagner-Schottkyģ�ͼ���Ԥ����Щ��ȱ�ݵ�Ũ�����¶Ⱥͳɷֵı仯��ϵ��������Щ��ȱ��֮��Ŀ�������ã�Ԥ���ȱ���γɼ����ܵĿ���ֵ��ΪL10-TiAl�����仯�����ʵ���Ʊ���ȱ�ݷ�������ѧ���ܸĽ����ṩ��������۲ο���
1 ���㷽�������
���о������м�����ڵ�һԭ��ƽ�沨���Ʒ���������������֮�������ò���ͶӰ�Ӳ�����(PAW)[18]����ȷ����������֮��Ľ��������Ʋ��ù����ݶȽ����µ�PBE����������[19]��Ti��3p4s3d��Al��3s3p������ΪTi��AlԪ�صļ۵��ӣ������ڲ������Ϊо���������ǡ�����ģ�ͼ�Լ����Ԩ��K���������Monkhorst-Pack[20]���������֣�ϵͳ�������ļ������BLOCHL��[21]������Linear-Tetrahedron����������Kohn-Sham��������ƽ�沨����չ��������Ŀ�ɾ��������Բ��Ժ�Ķ��ܽضϵ���ȷ�����ڼ��㾫��ȷ��Ϊ1��10-3 eV�������£�Ti��Al��TiAl���������Ķ��ܽضϵ�ȷ��Ϊ400 eV���������VASP(Vinena Ab-inito simulation package)���ܼ��������С��ڼ��㺬��ȱ�ݵľ����ṹʱ������һ������54��ԭ�ӵij��������ռ�����Ļ���ȡΪ4��4��3��ͼ1��ʾΪL10-TiAl�����仯���P���и��ֵ�ȱ�ݵij����ṹʾ��ͼ��
2 ������
2.1 Ti��Al��L10-TiAl�ľ��������嵯��ģ��
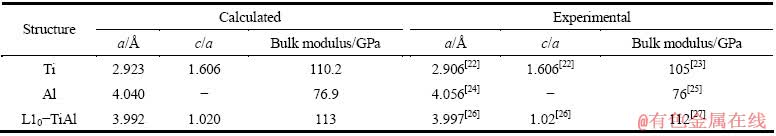
��1����Ϊ����õ���Ti��Al��L10-TiAl�����仯�������ṹ�����͵���ģ�������������[20-27]�е�ʵ��ֵ�Ƚϣ�����������������Ϊ0.58%(Ti)������ģ����Ϊ5%(Ti)�����۽����������ʵ�������ϽϺá�
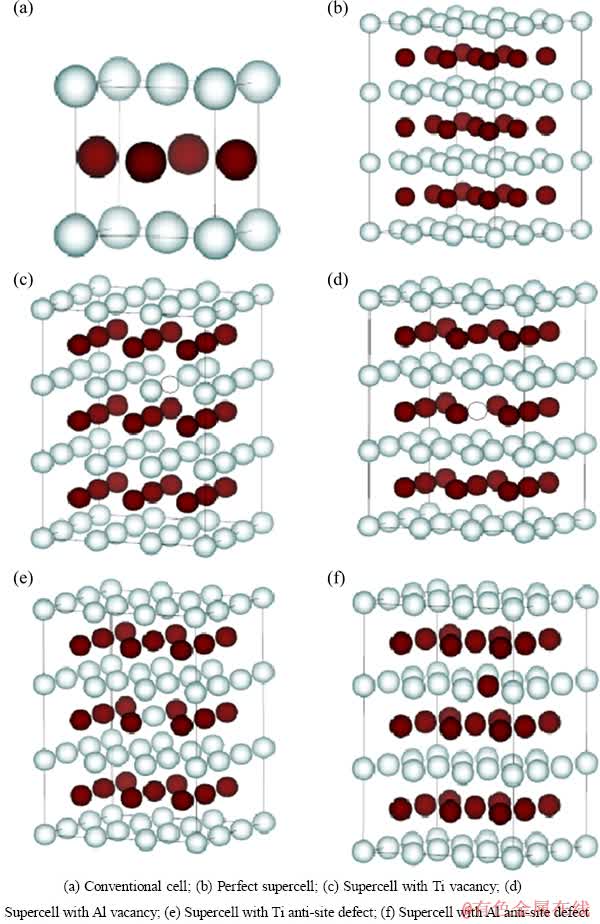
ͼ1 L10-TiAl�����仯�����λ�ͷ�λ��ȱ�ݾ���ģ��
Fig. 1 Supercell model of L10-TiAl intermetallics
��1 Ti��Al��L10-TiAl�ļ���ֵ��ʵ��ֵ�Ƚ�
Table 1 Comparison of calculated results with experimental data of Ti, Al, and L10-TiAl in comparison
2.2 L10-TiAl�ĵ�ȱ���γ���
����ȱ�ݵ�Ũ���㹻ϡʱ�����Բ���Wagner-Schottkyģ��[10]���о���ȱ��Ũ����ɷֺ��¶ȵı仯��ϵ�������ȱ��Ũ��ʱ���õ���ԭ��Ũ�ȵ���ʽ���ʵ�i(ԭ�ӻ��λ)�ڦ��ǵ���λ�õ�ԭ��Ũ�ȿɶ���Ϊ
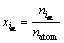 (1)
(1)
ʽ�У�natomΪ��ϵ��ԭ�ӵ�����(��������λ)��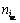 Ϊ�ʵ�i(ԭ�ӻ��λ)ռ�ݦ��ǵ���λ�õ���Ŀ����L10-TiAl�����仯�����У��ʵ�iΪ{Ti,Al,V}�����ǵ����Ϊ{Ti,Al}������V��ʾ��λ���ڵ�ȱ��(VAl��VTi��AlTi��TiAl)�У�VAl��ʾռ��Al�ǵ�����Ŀ�λ(Al��λ)��VTi��ʾռ��Ti�ǵ�����Ŀ�λ(Ti��λ)��AlTi��ʾռ��Ti�ǵ������Alԭ��(Al��λ)��TiAl��ʾռ��Al�ǵ������Tiԭ��(Ti��λ)������Wagner-Schottkyģ�ͣ�����ȱ�ݵ�L10-TiAl���γ������ȱ��Ũ�ȳ����ȣ��Ӷ���
Ϊ�ʵ�i(ԭ�ӻ��λ)ռ�ݦ��ǵ���λ�õ���Ŀ����L10-TiAl�����仯�����У��ʵ�iΪ{Ti,Al,V}�����ǵ����Ϊ{Ti,Al}������V��ʾ��λ���ڵ�ȱ��(VAl��VTi��AlTi��TiAl)�У�VAl��ʾռ��Al�ǵ�����Ŀ�λ(Al��λ)��VTi��ʾռ��Ti�ǵ�����Ŀ�λ(Ti��λ)��AlTi��ʾռ��Ti�ǵ������Alԭ��(Al��λ)��TiAl��ʾռ��Al�ǵ������Tiԭ��(Ti��λ)������Wagner-Schottkyģ�ͣ�����ȱ�ݵ�L10-TiAl���γ������ȱ��Ũ�ȳ����ȣ��Ӷ���
 (2)
(2)
ʽ�У���HTiAl�Ƿ��ϻ�ѧ�����ȵ���ȫ���������L10-TiAl���γ���(�Դ�����Ϊ�ο�̬)��xdΪ��ȱ��(VAl��VTi��AlTi��TiAl)��ԭ��Ũ�ȣ�HdΪ������ȱ����L10-TiAl�е�����γ���(������L10-TiAlΪ�ο�̬)��
��ȱ��L10-TiAl�Ͻ���γ��ʿɰ���ʽ���㣺
 (3)
(3)
ʽ�У�m��n�Ǿ���������ԭ����Ŀ��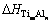 �Ǿ���
�Ǿ���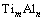 ���γ��ʣ�ETi��EAlΪ�������ô�����Ti��Alƽ����ÿ��ԭ�ӵ�������������ѧ�ơ�
���γ��ʣ�ETi��EAlΪ�������ô�����Ti��Alƽ����ÿ��ԭ�ӵ�������������ѧ�ơ�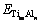 �Ǿ����������������У�����L10-TiAl���γ��ʿɰ����¹�ʽ���㣺
�Ǿ����������������У�����L10-TiAl���γ��ʿɰ����¹�ʽ���㣺
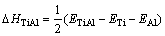 (4)
(4)
ʽ�У�ETiAl��L10-TiAl����������������
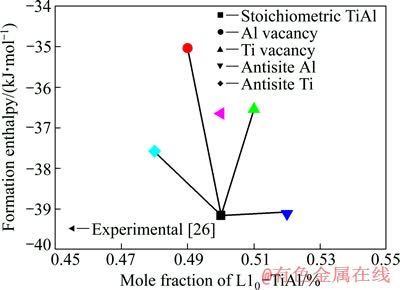
ͼ2 L10-TiAl�γ�����ɷ�֮��Ĺ�ϵ
Fig. 2 Relationship between of formation enthalpy and composition of L10-TiAl
����ʽ(3)��(4)�ɼ���õ�������ȱ��(VAl��VTi��AlTi��TiAl)��L10-TiAl���γ��ʣ������ͼ2��ʾ����������ѧ��֪���ϵ͵��γ�����ζ�Ÿ����ȶ���״̬����ͼ2���Կ���������λȱ��L10-TiAl���γ��ʱȿ�λȱ�ݵĸ��ͣ�������λȱ���������Ͽ�����L10-TiAl������ȶ���ȱ����ʽ�����ļ�����������L10-TiAl���γ����������б�����ʵ��ֵ������С��7%�����ϽϺá�
����Wagner-Schottkyģ����ȷ���Ĺ�ʽ(2)��ͼ2���γ��ʽ�����õ�������L10-TiAlΪ�ο�̬�ĵ�����ȱ��(VAl��VTi��AlTi��TiAl)�γ���Hd[10]��
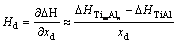 (5)
(5)
����õ��Ŀ�λ�ͷ�λȱ���γ��ʽ����������������ʵ��ֵ�Աȼ���2��
��2 L10-TiAl�����仯�����λ�ͷ�λ�γ���
Table 2 Calculated formation enthalpy of vacancies and anti-site defects in L10-TiAl
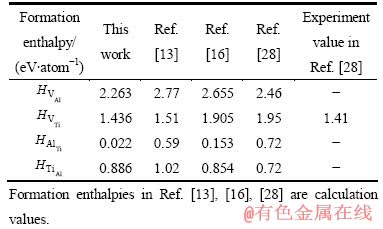
�ɱ�2��֪�����ļ�����������[13��16��28-29]�б������ļ����ʵ�������ϽϺã�ֻ��Al��λȱ�ݵ��γ��ʼ�����ƫ�͡����ͨ������Tiԭ��3s̬������Ϊ�۵��ӡ����K�������ܶȡ�����ƽ�沨���ܽضϵȷ������������¼��㣬������������Ըı䡣ͬʱ����2��ʾ�ı仯����������[13]��[16]һ�£���L10-TiAl��Ti��λȱ�ݵ��γ���ֵҪ����Al��λȱ�ݵģ�Al��λ���γ��ʴ���Ti��λ�ġ��ݴ˿��Թ��ƣ�����ƫ�����뻯ѧ�����ȵĸ�Al��TiAl��������ܵ���Ҫ�ṹȱ����ʽΪAl��λ��Ti��λ����ǰ�ߵ��γ���ԶС�ں��ߵģ�Al��λȱ�ݵ�Ũ�Ƚ�ռ�������ơ�ͬʱ�����ڸ�Ti��TiAl��������ܳ��ֵ���Ҫ��ȱ����ʽΪTi��λ��Al��λ����Ti��λȱ�ݵ�Ũ�Ƚ�ռ�������ơ���ˣ���ƫ�뻯ѧ�����ȵ�L10-TiAl�����仯�����У����Ǹ������γɷ�λȱ�ݣ���Ti���߸�Al��L10-TiAl���ֱ���Ti��λ����Al��λΪ��Ҫ��ȱ����ʽ�����ڡ�
2.3 L10-TiAl�ĵ�ȱ��Ũ��
�ڼ���TiAl�����仯�����ȱ��Ũ��ʱ��Ҫ�����¶ȵ����أ��Ҳ��ܺ����ض�ϵͳ��Ӱ�졣������������������̬�ص�Ӱ�죬����ͨ��ƽ����������������̬�أ�
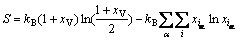 (6)
(6)
ʽ�У�kBΪ��������������xVΪ��λȱ�ݵ�Ũ�ȣ�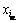 Ϊ��ʽ(1)�������ԭ��Ũ�ȡ���ʽ(6)���뼪��˹�����ܹ�ʽ��������˹�����ܴﵽ��Сֵʱ�ɵõ�ϵͳ���ȶ�̬��ƽ��̬����ʱ��Ӧ�ĵ�ȱ��Ũ�ȼ�Ϊƽ��Ũ�ȣ���ˣ�����ͨ����
Ϊ��ʽ(1)�������ԭ��Ũ�ȡ���ʽ(6)���뼪��˹�����ܹ�ʽ��������˹�����ܴﵽ��Сֵʱ�ɵõ�ϵͳ���ȶ�̬��ƽ��̬����ʱ��Ӧ�ĵ�ȱ��Ũ�ȼ�Ϊƽ��Ũ�ȣ���ˣ�����ͨ����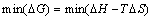 �õ�ϵͳ�����ȶ�̬ʱ�ĵ�ȱ��ƽ��Ũ�ȵ���Ԫ���η����飺
�õ�ϵͳ�����ȶ�̬ʱ�ĵ�ȱ��ƽ��Ũ�ȵ���Ԫ���η����飺
 (7)
(7)
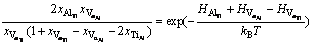 (8)
(8)
 (9)
(9)
 (10)
(10)
ͨ����ֵ���ʽ(7)~(10)���Եõ�L10-TiAl�����仯�����ȱ��Ũ�����¶ȼ��ɷֱ仯�Ĺ�ϵ����ͼ3��ʾ�����ǵ�L10-TiAl�������ɷ���Ҫ��0.48~0.55(Al��Ħ������)���䣬ͼ3��ʾΪ�óɷ�����ĵ�ȱ��Ũ�ȷֱ���973��1173��1373��1573 K�¶�����ɷֵı仯��ϵ��
��ͼ3��֪��1) ��ȱ�ݵ�Ũ�Ⱦ����¶����߶����ӣ�2) Al��λ��Ti��λŨ����Alԭ��Ũ�ȵ����Ӷ��½���Ti��λ��Al��λŨ����Alԭ��Ũ�ȵ����Ӷ����ӣ�3) ��ͬ�¶��£���λȱ�ݵ�Ũ�����Ǵ��ڿ�λȱ�ݵ�Ũ�ȣ�4) Ti��λŨ��ʼ�մ���Al��λŨ�ȣ����2��ȱ���γ��ʽ��һ�£���Ti��λ�γ��ʸ��ͣ��������γɣ�5) �ڻ�ѧ�����ȴ���Ti��λ��Al��λȱ��Ũ�Ȼ����൱�����ڸ�Ti��(xAl��50%)��Ti��λŨ�ȸ���Al��λ���ڸ�Al��(xAl��50%)���෴��
Ϊ��һ���о���ȱ��Ũ�����¶�֮��Ĺ�ϵ��ѡȡͼ3�����㻯ѧ��������������xAl=50%��L10-TiAlΪ�о������㵱�ɷ̶ֹ�ʱ��ȱ��Ũ�����¶ȱ仯�Ĺ�ϵ��������ͼ4��ʾ��

ͼ3 L10-TiAl�����仯����ĵ�ȱ��Ũ���ڲ�ͬ�¶�����ɷ�֮��Ĺ�ϵ
Fig. 3 Relationship between point defect concentrations and composition of L10-TiAl intermetallic composite at different temperatures
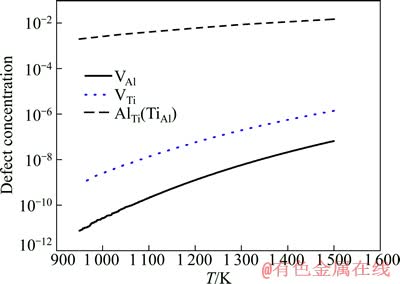
ͼ4 ���뻯ѧ������L10-TiAl��ȱ��Ũ�����¶ȵĹ�ϵ
Fig. 4 Relationship between point defect concentration and temperature in stoichiometric L10-TiAl
��ͼ4��֪��1) ��ȱ��Ũ�����¶����߶������ڽϵ����������Ͽ죬�ڽϸ��¶��������仺��2) Al��λȱ�ݺ�Ti��λȱ�ݵ�Ũ�ȼ�����ͬ���������ڿ�λŨ�ȣ�3) Ti��λ��Ũ�ȸ���Al��λ�ġ�
2.4 L10-TiAl��ȱ���γɼ����ܵ�ʵ��ֵԤ��
ͼ4��ʾΪ����Ԥ������뻯ѧ������L10-TiAl�в�ͬ��ȱ��Ũ�����¶ȵı仯���ơ���ʵ���о��У�ʵ�ʲⶨ��ȱ���γɼ�����ͨ��������Arrhenius���̣���
 (8)
(8)
ʽ�У�cΪ��ȱ��Ũ�ȣ�AΪƽ�ⳣ����QΪ��ȱ���γɼ�����(kJ��mol-1)��R��Ħ�����峣����TΪ����ѧ�¶ȡ���ʽ(8)ȡ�������ɵ�
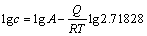 (9)
(9)
��Ȼ��lgc��1/T�������Թ�ϵ������T�ɷ��ȹ�ϵ���ı�ͼ4�ĺ�����ΪT�ĵ���(T -1)��ȡ��ȱ��Ũ��cΪ�����꣬���»������뻯ѧ������L10-TiAl�е�ȱ��Ũ�����¶ȹ�ϵ���õ������ͼ5��ʾ��
ͨ����ϸ�ֱ�ߵ�б�ʣ��ɵõ����뻯ѧ������L10-TiAl�в�ͬ��ȱ��(VAl��VTi��AlTi��TiAl)��Ӧ���γɼ�����(eV��atom-1)������㹫ʽ���£�
 (10)
(10)
ʽ�У�kBΪ�����������������������3���С��ɱ�3��֪��Al��λȱ�ݵ��γɼ��������Ti��λȱ�ݵĴ�֮��Al��λȱ�ݺ�Ti��λȱ�ݵ��γɼ�������С���൱����3��Ԥ����γɼ�����ʵ������һ�ֱ��ۼ����ܣ���ֱ����ʵ����ֱ�ӶԱȣ���Ϊʵ����֤���ǵ����ۼ������ṩ��һ�ֿ��ܡ�
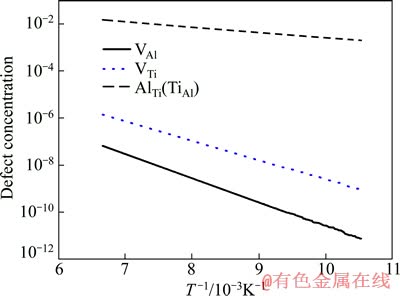
ͼ5 L10-TiAl�����仯�����ȱ��Ũ�����¶�֮��Ĺ�ϵ
Fig. 5 Relationship between point defect concentrations with temperature intermetallics composite of L10-TiAl
��3 L10-TiAl�п�λ�ͷ�λȱ���γɼ����ܵ�ʵ��ֵԤ��
Table 3 Prediction of activation energy of vacancy and anti-site defects in intermetallics composite L10-TiAl (eV��atom-1)

2.5 L10-TiAl��ȱ�ݶԵ��γ���
���о������˵�����ȱ��(VAl��VTi��AlTi��TiAl)���γ��ʣ�����Wagner-Schottkyģ���о���ȱ��Ũ�����¶Ⱥͳɷֵı仯����Ԥ���γɼ����ܵ�ʵ�����ֵ���ڴ˻����ϣ������о���Щ��ȱ��֮��Ŀ�������á��ֽ���Щ��ȱ�ݽ�����ϣ��õ�6�ֿ��ܵ�ȱ�ݶ�(d-d��)�ṹ����VAl-VTi��VAl-AlTi��VTi-TiAl��AlTi-TiAl��VAl-TiAl��VTi-AlTi��ͼ6��ʾΪ��ȱ�ݶԵij���ģ�ͣ�������Щģ�ͼ�����Щ��ȱ�ݶԵ������������뵥����ȱ�ݽ��������Աȣ�������������������L10-TiAlΪ�ο�̬���γ��ʺͽ���ܣ��Ӷ�Ԥ������֮����������ų�����[30]��
��˫��λ��ȱ�ݶ�(VAl-VTi)Ϊ�������γ��ʺͽ���ܼ��㹫ʽ����[31]��
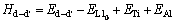 (11)
(11)
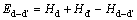 (12)
(12)
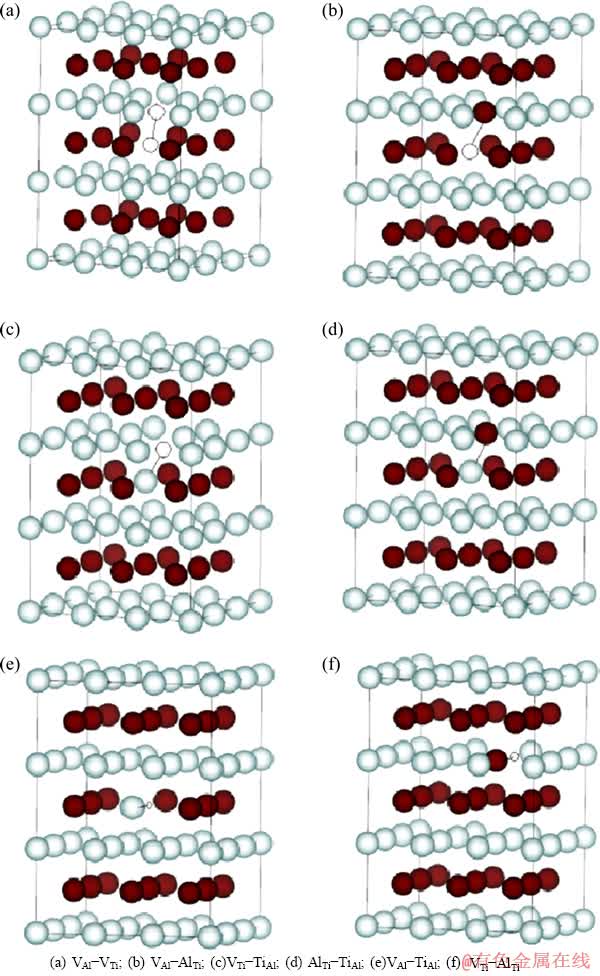
ͼ6 L10-TiAl��ȱ�ݶԵľ���ģ��
Fig. 6 Supercell model of point defect pairs in L10-TiAl
ʽ(11)�е�EAl��ETi��ʽ(3)�еĶ�����ͬ���ֱ��ʾ��Ti�ʹ�Al�Ļ�ѧ�ơ�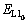 ��ʾ���뻯ѧ�����ȵ�L10-TiAl������������
��ʾ���뻯ѧ�����ȵ�L10-TiAl������������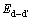 ��ʾ����ȱ�ݶ�(d-d��)�ij�����������
��ʾ����ȱ�ݶ�(d-d��)�ij�����������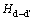 Ϊ��ȱ�ݶԵ��γ��ʡ�ʽ(12)�У���ʾȱ�ݶ�(d-d��)�Ľ���ܣ�Hd��
Ϊ��ȱ�ݶԵ��γ��ʡ�ʽ(12)�У���ʾȱ�ݶ�(d-d��)�Ľ���ܣ�Hd��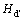 Ϊ������ȱ�ݵ��γ���(����2)�����м������ɼ���4��
Ϊ������ȱ�ݵ��γ���(����2)�����м������ɼ���4��
�ɱ�4��֪��1) ��ȱ�ݶ��γ��ʵĸߵ�˳������ΪVAl-VTi��VAl-TiAl��VAl-AlTi��VTi-TiAl��VTi-AlTi��AlTi-TiAl������AlTi-TiAlȱ�ݶԵ��γ���ֵ��ͣ���Զ������ȶ���2) ���е�ȱ�ݶԵĽ���ܾ�Ϊ��ֵ������L10-TiAl�е���Щ��ȱ�����Ծۼ�������������������ɢ����ɢ��
��4 L10-TiAl�е�ȱ�ݶԵ��γ��ʺͽ����
Table 4 Formation enthalpies and binding energies of point defect pairs in L10-TiAl
3 ����
1) L10-TiAl�����仯���������б�����ȱ�ݵ��γ��ʾ�Ϊ��ֵ������Al��Ti��λȱ�ݵľ���ֵ��������λȱ����L10-TiAl����Ϊ�ȶ�����ȱ��Ũ�Ⱦ����¶����߶����ӣ��ҷ�λȱ��Ũ��ʼ�մ��ڿ�λȱ��Ũ�ȣ������ڲ�ͬ�¶��£���λȱ��ʼ����L10-TiAl������Ҫ�ĵ�ȱ����ʽ��
2) ������L10-TiAl�����ɷַ�Χ��(Al����Ϊ48%~55%��Ħ������)��Al��λ��Ti��λȱ��Ũ�Ⱦ���Al�������Ӷ��½�����Ti��λ��Al��λŨ�ȱ仯�����෴�������뻯ѧ������(AlΪ50%)����Ti��λ��Al��λȱ��Ũ�Ȼ����൱�����ڸ�Ti�ˣ�Ti��λŨ�ȸ���Al��λŨ�ȣ��ڸ�Al�ˣ�Al��λŨ�ȸ���Ti��λŨ�ȡ�
3) ����Arrhenius���̼���Ԥ����ֵ�ȱ�ݵ��γɼ����ܣ�����Al��λ�γɼ��������Ti��λ��֮��Al��λ��Ti��λ�γɼ����ܽ�С����ֵ�൱��ͨ���Բ�ͬ��ȱ��֮�������ļ��㿼�죬���ֵ�ȱ��֮���������ų��ԣ����Ծۼ�������������������ɢ����ɢ��
��л����л���ϴ�ѧ�����ܼ�������Ϊ���м��㹤���ṩӲ������֧�֡�
REFERENCES
[1] �� ��, �����. �����仯������½ṹ���ϵ��о�����[J]. ���ϵ���, 1994(4): 14-18.
CAO Yang, LI Guo-jun. The recent development of high-temperature structural intermetallics[J]. Materials Review, 1994(4): 14-18.
[2] �ܻ�Ӫ, տ����. TiAl�����仯������о���չ[J]. ������ѧѧ��, 1999, 24(4): 262-264.
ZHOU Huai-ying, ZHAN Yong-zhong. Development of studies on TiAl intermetallics[J]. Journal of Guangxi University, 1999, 24(4): 262-264.
[3] ����, Ԭ����, �ܾ���, ��־��. TiAl���Ͻ��о���չ[J]. �������켼��, 2009(3): 35-38.
FENG Xu-dong, YUAN Qing-long, CAO Jing-jing, SU Zhi-jun. Progress in TiAl-based alloys[J]. Aerospace Manufacturing Technology, 2009(3): 35-38.
[4] ���ɽ, ������, �� ��, �ܺ곬, �� ��. TiAl�������仯������о���״�뷢չ����[J]. �й����Ͻ�չ, 2010, 29(3): 1-5.
LI Jin-shan, ZHANG Tie-bang, CHANG Hui, KOU Hong-chao, ZHOU Lian. Recent achievements and future directions of TiAl based intermetallic compounds[J]. Materials China, 2010, 29(3): 1-5.
[5] �� ��, �� �, �� ��. �����仯����ṹ���Ϸ�λȱ�ݼ�������ܵ�Ӱ��[J] .ϡ�н��������빤��, 2013, 42(2): 429-434.
ZHANG Jing, CHEN Zheng, YANG Tao. Antisite defect in the intermetallic structural materials and its effect on the mechanical performance[J]. Rare Metal Materials and Engineering, 2013, 42(2): 429-434.
[6] ����ӱ, ������. ��ȱ�ݶԦ�-TiAl(100)����Oԭ����������ɢӰ��ĵ�һ��ԭ���о�[J]. ����ѧ��, 2013, 49(11): 1387-1391.
ZHOU Li-ying, WANG Fu-he. First-principles study of effect of point defect on adsorption and diffusion of oxygen at ��-TiAl (100) surface[J]. Acta Metallurgica Sinica, 2013, 49(11): 1387-1391.
[7]  U, APPEL F.Strain ageing in ��(TiAl)-based titanium aluminides due to antisite atoms[J]. Acta Materialia, 2002, 50: 3693-3707.
U, APPEL F.Strain ageing in ��(TiAl)-based titanium aluminides due to antisite atoms[J]. Acta Materialia, 2002, 50: 3693-3707.
[8] JIANG C, CHEN L Q, LIU Z K.First-principles study of constitutional point defects in B2 NiAl using special quasirandom structures[J]. Acta Materialia, 2005, 53: 2643-2652.
[9] JIANG C. Site preference of early transition metal elements in C15 NbCr2[J]. Acta Materialia, 2007, 55(5): 1599-1605.
[10] ��˳ƽ, ��Сƽ, �� �S, ¬����, � ��, ����, ����. L12-Al3Li�����仯�����ȱ��Ũ�ȵĵ�һԭ������[J]. �й���ɫ����ѧ��, 2013, 23(2): 370-378.
SUN Sun-ping, LI Xiao-ping, YU Yun, LU Ya-lin, ZANG Bing, YI Dan-qing, JIANG Yong. First-principle calculation of point defects concentration in L12-Al3Li intermetallic[J]. The Chinese Journal ofNonferrousMetals, 2013, 23(2): 370-378.
[11] KORZHAVYI P A, RUBAN A V, LOZOVOI A Y, VEKILOV Y K, ABRIKOSOV I A, JOHANSSON B. Constitutional and thermal point defects in B2 NiAl[J]. Physical Review B, 2000, 61(9): 6003-6018.
[12] ZHU G L,DAI Y B, SHU D, XIAO Y P, YANG Y X, WANG J, SUN B D, BOOM R. First-principles study of point defects and Si site preference in Al3Ti[J]. Computational Materials Science, 2011, 50 (9): 2636-2639.
[13] �� ��, �� ƽ, ������. L10-TiAl�������Եļ�����Ƚ��о�[J]. ���Ͽ�ѧ�빤��, 2007, 15(1): 47-51.
CHEN L��, PENG Ping, HAN Ya-li. A comparison on basic physical properties of L10-TiAlcalculatedby first-principles methods[J] MaterialsScienceandTechnology, 2007, 15(1): 47-51.
[14] �� ��. 3d���ɽ�����L10-TiAl��ռλ�ĵ�һԭ������[J]. ��ɳ����ְҵ����ѧԺѧ��, 2008, 8(3): 43-48.
CHEN L��. First-principles calculation for site substitution of 3d transition metal elements in L10-TiAl intermetalliccompound[J]. Journal of Changsha Aeronautical Vocational and Technical College, 2008, 8(3): 43-48.
[15] JIANG C. First-principles study of site occupancy of dilute 3d, 4d and 5d transition metal solutes in L10 TiAl[J]. Acta Materialia, 2008, 56(20): 6224-6231.
[16] LI Yu-juan, HU Qing-miao, XU Dong-sheng, YANG Rui. Strengthening of g-TiAl-Nb by short-range ordering of point defects[J]. Intermetallics, 2011, 19: 793-796.
[17] WANG H, REED R C, GEBELIN J C, WARNKEN N. On the modelling of the point defects in the ordered B2 phase of the Ti-Al system: Combining CALPHAD with first-principles calculations[J]. Calphad: Computer Coupling of Phase Diagrams and Thermochemistry, 2012, 39: 21-26.
[18] KRESSE G, JOUBERT J. From ultrasoft pseudopotentials to the projector augmented-wave method[J]. Physical Review B, 1999, 59(3): 1758-1775.
[19] PERDEW J P, BURKE K M, EMZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letters, 1996, 77(18): 3865-868.
[20] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13(12): 5188-5192.
[21] BLOCHL P E, JEPSEN O, ANDERSEN O K. Improved tetrahedron method for Brillouin-zone integrations[J]. Physical Review B, 1994, 49(23): 16223-16233.
[22] NOVOSELOVA T, MALINOV S, SHA W, ZHECHEVA A. High-temperature synchrotron X-ray diffraction study of phases in a gamma TiAl alloy[J]. Materials Science and Engineering A, 2004, 371(1): 103-112.
[23] ������, �� ��, �ﶫ��, �� ��, �� ǿ. ����Ѿ��嵯��ģ��Ӱ��ĵ�һԭ���о�[J]. ���Ͽ�ѧ�빤��, 2009, 17(3): 305-310.
HAN Xiu-li, WANG Qing, SUN Dong-li, SUN Tao, GUO Qiang. First-principles study of the effect of hydrogen on the elastic moduli of titaniumcrystals[J]. Materials Science and Technology, 2009, 17(3): 305-310.
[24] TOUGAIT O,  H. Stoichiometry of UAl4[J]. Intermetallics, 2004, 12(2): 219-223.
H. Stoichiometry of UAl4[J]. Intermetallics, 2004, 12(2): 219-223.
[25] SEITZ F, TURNBU D. Solid state physics: Advance in research and applications(Volume 16)[M]. New York: Academic Press, 1964.
[26] ZOPE R R, MISHIN Y. Interatomic potentials for atomistic simulations of the Ti-Al system[J]. Physical Review B, 2003, 68(2): 02402.
[27] DENIS M, SCHNEIDER J M. Effect of transition metal additives on electronic structure and elastic properties of TiAl and Ti3Al[J]. Physical Review B, 2006, 74(17): 174110-174114.
[28] YOO M H, FU C L. Physical constants, deformation twinning, and microcracking of titanium aluminides[J]. Metallurgical and Materials Transactions A, 1998, 29(1): 49-63.
[29] BROSSMANN U, W��RSCHUM R, BADURA K, SCHAEFER H E. Thermal formation of vacancies in TiAl[J]. Physical Review B, 1994, 49(10): 6457-6466.
[30] FU C L, WANG X D. The effect of electronic structure on the defect properties of FeAl[J]. Materials Science and Engineering A, 1997, 239/240: 761-768.
[31] PARLINSKI K, JOCHYM P T, KOZUBSKI, ORAMUS P. Atomic modelling of Co, Cr, Fe, antisite atoms and vacancies in B2�CNiAl[J]. Intermetallics, 2003, 11(2): 157-160.
(�༭ ����)
������Ŀ������ʡ�Ƽ��ƻ���Ŀ(2013GK3010)�����һ����о���չ�ƻ�������Ŀ(2014CB644001-2)
�ո����ڣ�2014-05-13�������ڣ�2014-09-11
ͨ�����ߣ��� �£����ڣ���ʿ���绰��15111309497��E-mail��yjiang@csu.edu.cn

