J. Cent. South Univ. Technol. (2011) 18: 998-1003
DOI: 10.1007/s11771-011-0793-4
First principles study on electronic structure and optical properties of quaternary arsenide oxides YZnAsO and LaZnAsO
SHI Yi-ming(施毅敏)1, YE Shao-long(叶绍龙)2
1. Department of Basic Teaching, Hunan Institute of Technology, Hengyang 421000, China;
2. School of Metallurgical Science and Engineering, Central South University, Changsha 410083, China
? Central South University Press and Springer-Verlag Berlin Heidelberg 2011
Abstract: The electronic structure and optical properties of the tetragonal phase quaternary arsenide oxides YZnAsO and LaZnAsO were studied using density-functional theory (DFT) within generalized gradient approximation (GGA). The band structure along the higher symmetry axes in the Brillouin zone, the density of states (DOS) and the partial density of states (PDOS) were presented. The calculated energy band structures show that both YZnAsO and LaZnAsO are indirect gap semiconductors with band gap of 1.173 1 eV and 1.166 5 eV, respectively. The DOS and PDOS show the hybridization of Y-O/La-O atom orbits and Zn-As atom orbits. The dielectric function, reflectivity, absorption coefficient, refractive index, electron energy-loss function and optical conductivity were presented in an energy range from 0 to 25 eV for discussing the optical properties of YZnAsO and LaZnAsO.
Key words: YZnAsO/LaZnAsO; density-functional theory; generalized gradient approximation; electronic structure; optical properties
1 Introduction
The quaternary oxypnictides layered compounds REZnPO (RE=Y, La-Nd, Sm, Gd, Dy, Ho) and REZnAsO (RE=Y, La-Nd, Sm, Gd-Er) with the ZrCuSiAs-type structure have received considerable attention [1-6] because of the discovery of superconductivity in LaOFeP, LaNiOP and LaFeAsO(F) [7-11]. They have similar layered structures with alternating [REO] and [ZnPn] (Pn=As, P) layers, and exhibit a wide variety of interesting electrical, optical, and magnetic properties [1-6]. YZnAsO and LaZnAsO, which are isostructural to the layered rare-earth oxypnictides REZnAsO (RE=Y, La-Nd, Sm, Gd, Er), are semiconductor compounds with band gap about 1.5- 2.3 eV in experimental investigation [2, 5] or about 0.65-1.3 eV in theoretical calculation [6]. KAYANUMA et al [2] have successfully grown LaZnAsO thin films on MgO substrates. The electrical conductivity of the undoped LaZnOAs epitaxial films is 2.0×10-1 S/cm at room temperature, the Seebeck coefficient of the epitaxial film is positive and the optical bandgaps of LaZnOAs is estimated to be ~1.5 eV, indicating that LaZnAsO is a p-type semiconductor and may be a promising matrix for diluted magnetic semiconductors if Zn2+ is partially replaced by a divalent transition metal, for example Mn2+. LINCKE et al [5] have synthesized well crystallized form REZnAsO (RE=Y, La) from the ingots of rare earth elements, arsenic, and ZnO in NaCl/ KCl fluxes in sealed silica ampoules. The REZnAsO structures consist of alternate stacks of (RE3+O2-) and (Zn2+As3-) layers with covalent RE-O and Zn-As intralayer and weak ionic interlayer bonding. LaZnAsO and YZnAsO show an optical bandgap of ~2.3 eV and ~1.9 eV, respectively. BANNIKOV et al [6] have investigated the electronic properties and chemical bonding picture of LaZnAsO and YZnAsO. The band structure results show that both of them are semiconductors with the band gaps at about 0.65 eV for LaZnAsO and 1.3 eV for YZnAsO. The bonding can be classified as a mixture of ionic and covalent contributions. To the best of our knowledge, there are no detail reports of optical properties of the tetragonal phase quaternary arsenide oxides YZnAsO and LaZnAsO. However, it is important for fundamental physics and potential applications to study optical properties of YZnAsO and LaZnAsO. In this work, the electronic structures and optical properties of YZnAsO and LaZnAsO were studied using first principles calculations based on DFT. First, the electronic structures were carefully calculated, because the optical properties depend on both the inter-band and intra-band transitions determined by the energy bands. Then, the optical properties, including the dielectric function, the reflectivity, the electron energy-loss function, the absorption coefficient, the optical conductivity and the refractive index were discussed.
2 Calculation details and models
The first principle calculations described here are based on DFT using a plan-wave expansion of the wave function [12-13]. The exchange correlation energy was calculated by the GGA with the Perdew-Burke- Ernzerhof (PBE) function [14]. The norm-conserving pseudopotential [15-17] was used for electron-ion interaction, which can give a more accurate description of optical properties than ultrasoft pseudopotentials [16-17]. The ionic cores were represented by norm-conserving pseudopotentials for Y, La, O, Zn and As atoms. The Y 4d15s2 electrons, La 5d16s2 electrons, O 2s22p4 electrons, Zn 3d104s2 electrons, and As 4s24p3 electrons were explicitly treated as valence electrons. The Monkhorst and Pack scheme of k-point sampling was used for integration over the first Brillouin zone [18]. The cutoff energy was chosen to be 900 eV, and the Brillouin-zone sampling k-point set mesh parameters were 12×12×6. This set of parameters assure the total energy convergence of 5.0×10-6 eV/atom, the maximum force of 0.01 eV/?, the maximum stress of 0.02 GPa and the maximum displacement of 5.0×10-4 ?.
Finally, within the electric-dipole approximation, the imaginary parts of the dielectric functions ε2(ω) can be calculated from the momentum matrix elements between the occupied and unoccupied wave functions within the selection rules, and the real part of dielectric function ε1(ω) can be evaluated from the imaginary part of dielectric function by Kramer-Kr?nig relationship [19-20]. Therefore, all solid macroscopical optical constant, such as reflectivity R(ω), energy-loss function L(ω), optical absorption coefficient I(ω), optical conductivity σ(ω), refractive index n(ω) and extinction coefficient k(ω), can be calculated and analyzed using the imaginary part of dielectric function ε2(ω) and the real part of dielectric function ε1(ω). All the calculations were performed using CASTEP code [13].
3 Results and discussion
3.1 Geometry and structure properties
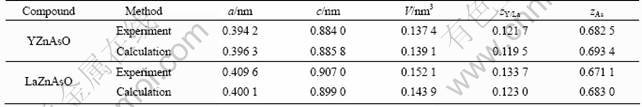
The crystal structure of the tetragonal phase of YZnAsO/LaZnAsO belongs to the space group P4/nmm, Z=2 (ZrCuSiAs type), where blocks [YO/LaO] are sandwiched with [ZnAs] blocks as depicted in Fig.1. There are four inequivalent atomic positions: Y/La at 2c site (1/4, 1/4, zY/La), O at 2a site (3/4, 1/4, 0), Zn at 2b site (3/4, 1/4, 1/2), and As at 2c site (1/4, 1/4, zAs) [2, 5-6] (zY/La and zAs are the internal coordinates of Y/La and As, respectively). The experimental lattice parameters are a=b=0.394 2/0.409 6 nm and c=0.884 0/ 0.907 0 nm, and the internal coordinates of Y/La and As, are reported as zY/La=0.121 7/0.133 7 and zAs=0.682 5/ 0.671 1 for YZnAsO and LaZnAsO [2, 5-6], respectively.
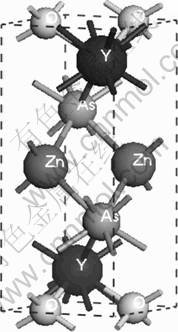
Fig.1 Crystal structure sketch of YZnAsO
In the first stage, the full structural optimization of this phase was performed both over the lattice parameters and at the atomic positions including the internal coordinate zY/La and zAs. The calculated optimization lattice parameters a (nm), c (nm), V (nm3) and atomic coordinates compared with available experimental data [2, 5] for YZnAsO and LaZnAsO are summarized in Table 1, which show that the calculated values of GGA calculation are in agreement with the experiments.
Table 1 Calculated lattice parameters a, c, V and atomic internal coordinate zY/La, zAs compared with experimental data [5] for YZnAsO and LaZnAsO
3.2 Electronic structure
The calculated energy band structure of YZnAsO and LaZnAsO along with the high-symmetry points of the Brillouin zone by GGA are shown in Fig.2. The top of the valence band is taken as the zero of energy. The compounds are found to have indirect band gap. The valence band maximum (VBM) is at the Γ point for both compounds and the conduction band minimum (CBM) is on the M point for YZnAsO and M-Γ line for YZnAsO. The calculated band gap values are 1.173 1 eV for YZnAsO and 1.166 5 eV for LaZnAsO, which are smaller than the corresponding experimental values 1.9 eV and 1.5 eV (2.3 eV) [2, 5] due to the well-known underestimation of conduction band state energies in DFT calculations.
The total density of states (TDOS) and partial density of states (PDOS) for YZnAsO and LaZnAsO are shown in Fig.3. There are five sub-bands in the whole calculated energy range. For YZnAsO, it is found that the lower valence bands (between -17.90 eV and -16.91 eV) are essentially dominated by O 2s states and show hybridization with Y 5s, Y 5p and Y 4d states. The structure situated in the range from -11.72 eV to -9.99 eV originates predominantly from As 4s, and the range from -8.50 eV to -7.23 eV originates pre- dominantly from Zn 3d states. The upper valence bands are mainly composed of Y 4d, O 2p and As 4p charac- teristics, and show hybridization of Y 4d with O 2p states and As 4p with Zn 4s, Zn 4p states. The conduction bands are contributed from all the atoms and with mainly Y 4d states hybridized with O 2s, O 2p states. The TDOS and PDOS for LaZnAsO have similar characteristic. All the calculated results are in agreement with the previous results [6].
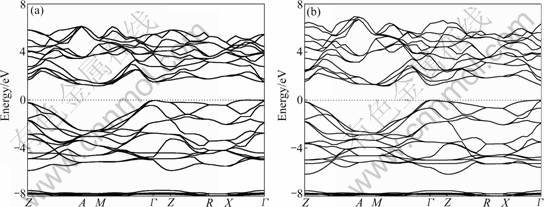
Fig.2 Calculated band structure of YZnAsO (a) and LaZnAsO (b) along some high-symmetry lines in Brillouin zone (Zero of energy is at Fermi level)
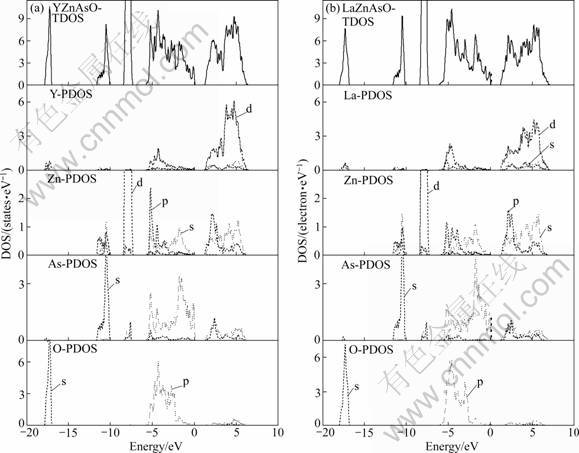
Fig.3 TDOS and PDOS of YZnAsO (a) and LaZnAsO (b)
3.3 Optical properties
Figures 4 and 5 show the optical functions of YZnAsO and LaZnAsO calculated for photon energies up to 25 eV. The study on the optical functions may help to give a better understanding of the electronic structure. The dielectric function curves as functions of the photon energy are displayed in Fig.4, where the solid line and dashed line represent the real part ε1(ω) and imaginary part ε2(ω), respectively. The calculated static dielectric function ε1(0) is 10.92 for YZnAsO and 10.16 for LaZnAsO. The real part dielectric function ε1(ω) of YZnAsO reaches two peak values at 2.16 eV and 3.18 eV, while for LaZnAsO, the two peak values are at 2.43 eV and 3.26 eV, and they are mainly generated by the electron transition from the top of the valence band to the bottom of conduction band. It is well known that the imaginary part dielectric function ε2 is the fundamental factor of the optical properties for any material. From the imaginary part dielectric function ε2 as a function of photon energy in Fig.4, it can be seen that the absorption starts at about 1.22 eV for YZnAsO and 1.16 eV for LaZnAsO, which is related to the minimum band gap near the Fermi level.
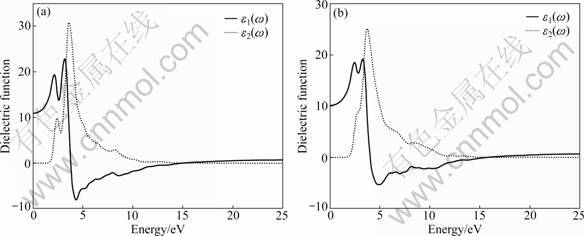
Fig.4 Dielectric function of YZnAsO (a) and LaZnAsO (b)
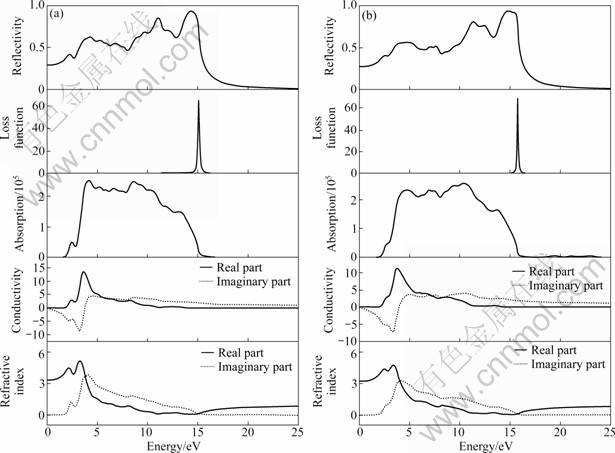
Fig.5 Reflectivity, loss function, absorption coefficient, optical conductivity and refractive index of YZnAsO (a) and LaZnAsO (b) as function of photon energy
Figure 5 shows the reflectivity, energy-loss function, absorption coefficient, optical conductivity and refractive index of YZnAsO and LaZnAsO as functions of photon energy. It is noticed that there are several reflectivity peaks in the calculated photon energies up to 25 eV for the two compounds. The reflectivity reaches the maximum value 93% at about 14.31 eV for YZnAsO and 94% at 14.86 eV for LaZnAsO, then drops off to about 50% at 15.25 eV and 15.93 eV.
L(ω) is an important factor describing the energy loss of a fast electron traversing in a material. The peaks on L(ω) spectra represent the characteristic associated with the plasma resonance and the corresponding frequency is the so-called plasma frequency. In addition, the peaks of L(ω) also correspond to the trailing edges in the reflection spectra [19-20]. The peaks of L(ω) for YZnAsO and LaZnAsO are at 15.07 eV and 15.75 eV, corresponding to the abrupt reduction of R(ω).
Absorption coefficient is a percentage that tells the decay of light intensity spreads in unit distance in medium. From the calculated absorption coefficients for YZnAsO and LaZnAsO, it can be seen that the absorption to light of YZnAsO and LaZnAsO is almost zero when the energy is below 1.55 eV. When the photon energy is larger than the value, the absorption coefficient will increase, which corresponds to the calculated indirect band gap of 1.173 1 eV and 1.166 5 eV of YZnAsO and LaZnAsO, respectively. There are many peaks within the energy range studied and the peak structures can be explained from the interband transitions using the band structure results stated above.
The imaginary part and real part of the optical conductivity are presented. The real part of the optical conductivity is zero until the photo energy increases to about 1.41 eV for YZnAsO and 1.33 eV for LaZnAsO, respectively. This is in correspondance with the band gaps of the band structure, DOS and PDOS calculations.
According to the relation of refractive index and dielectric function n2-k2=ε1 and 2nk=ε2, the refractive index n and extinction coefficient k of YZnAsO and LaZnAsO are obtained. The static refractive index n0 is 3.305 for YZnAsO and 3.188 for LaZnAsO, which agrees with the calculated results in Fig.4, where the corresponding calculated static dielectric function values ε1(0) are 10.92 and 10.16 (from 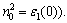 Extinction coefficient k indicates the absorption of light and at the same time, it also shows a great absorption characteristic at band-edge. The interband transitions discussed above also explain the origin of the peak structures in the refractive index and the extinction coefficient.
Extinction coefficient k indicates the absorption of light and at the same time, it also shows a great absorption characteristic at band-edge. The interband transitions discussed above also explain the origin of the peak structures in the refractive index and the extinction coefficient.
To the best of our knowledge, there are no detail experimental results on the optical properties of YZnAsO and LaZnAsO. We therefore hope that our calculations will motivate experimental studies in these compounds. Availability of experimental data will help in making quantitative comparisons with theoretical results.
4 Conclusions
1) The calculated energy band structures show that both YZnAsO and LaZnAsO are indirect gap semiconductors with band gap of 1.173 1 eV and 1.166 5 eV, respectively, which are smaller than the corresponding experimental values 1.9 eV and 1.5 eV (2.3 eV) due to the well-known underestimation of conduction band state energies in DFT calculations.
2) The DOS and PDOS of YZnAsO and LaZnAsO are presented, which show strong hybridization of Y PDOS/La PDOS states with O PDOS states and As PDOS states with Zn PDOS states.
3) The dielectric function, reflectivity, energy-loss function, absorption coefficients, optical conductivity, refractive index and extinction coefficient of YZnAsO and LaZnAsO were calculated. The relationship of the optical properties with the band structure and DOS (PDOS) were discussed.
4) The detailed optical properties of YZnAsO and LaZnAsO await experimental findings for comparison.
References
[1] KAYANUMA K, HIRAMATSU H, HIRANO M, KAWAMURA R, YANAGI H, KAMIYA T, HOSONO H. Apparent bipolarity and Seebeck sign inversion in layered semiconductor: LaZnOP [J]. Phys Rev B, 2007, 76: 195325.
[2] KAYANUMA K, KAWAMURA R, HIRAMATSU H, YANAGI H, HIRANO M, KAMIYA T, HOSONO H. Heteroepitaxial growth of layered semiconductors, LaZnOPn (Pn=P and As) [J]. Thin Solid Films, 2008, 516: 5800-5804.
[3] TAKANO Y, KOMATSUZAKI S, KOMASAKI H, WATANABE T, TAKAHASHI Y, TAKASE K. Electrical and magnetic properties of LnOZnPn (Ln= rare earths; Pn=P, As, Sb) [J]. J Alloys Compd, 2008, 451: 467-469.
[4] LINCKE H, GLAUM R, DITTRICH V, TEGEL M, JOHRENDT D, HERMES W, M?LLER M H, NILGES T, P?TTGEN R. Magnetic, optical, and electronic properties of the phosphide oxides REZnPO (RE=Y, La-Nd, Sm, Gd, Dy, Ho) [J]. Z Anorg Allg Chem, 2008, 634: 1339-1348.
[5] LINCKE H, GLAUM R, DITTRICH V, M?LLER M H, P?TTGEN R. Structure and optical properties of the arsenide oxides REZnAsO (RE=Y, La_Nd, Sm, Gd_Er) [J]. Z Anorg Allg Chem, 2009, 635: 936-941.
[6] BANNIKOV V V, SHEIN I R, IVANOVSKII A L. Electronic properties and chemical bonding in quaternary arsenide oxides LaZnAsO and YZnAsO [J]. Mater Chem Phys, 2009, 116: 129-133.
[7] KAMIHARA Y, HIRAMATSU H, HIRANO M, KAWAMURA R, YANAGI H, KAMIYA T, HOSONO H. Iron-based layered superconductor: LaOFeP [J]. J Am Chem Soc, 2006, 128: 10012-10013.
[8] WATANABE T, YANAGI H, KAMIYA T, KAMIHARA Y, HIRAMATSU H, HIRANO M, HOSONO H. Nickel-based oxyphosphide superconductor with a layered crystal structure, LaNiOP [J]. Inorganic Chemistry, 2007, 46(19): 7719-7722.
[9] KAMIHARA Y, WATANABE T, HIRANO M, HOSONO H. Iron-based layered superconductor La[O1-xFx]FeAs (x=0.05-0.12) with Tc=26 K [J]. J Am Chem Soc, 2008, 130: 3296-3297.
[10] WEN Hai-hu. Developments and perspectives of iron-based high-temperature superconductors [J]. Adv Mater, 2008, 20(19): 3764-3773.
[11] REN Zhi-an, ZHAO Zhong-xian. Research and prospects of iron-based superconductors [J]. Adv Mater, 2009, 21(45): S4584-S4595.
[12] PENG F, CHEN D, YANG X D. First-principles calculations on elasticity of OsN2 under pressure [J]. Solid State Commun, 2009, 149: 2135-2138.
[13] SEGALL M D, LINDAN PHILIP J D, PROBERT M J, PICKARD C J, HASNIP P J, CLARK S J, PAYNE M C. First-principles simulation: Ideas, illustrations and the CASTEP code [J]. J Phys: Condens Matter, 2002, 14(11): 2717-2744.
[14] PERDEW J P, BURKE K, ERNZERHOF M. generalized gradient approximation made simple [J]. Phys Rev Lett, 1996, 77: 3865-3868.
[15] HAMANN D R, SCHLUTER M, CHIANG C. Norm-conserving pseudopotentials [J]. Phys Rev Lett, 1979, 43: 1494-1497.
[16] ZHANG X Y, CHEN Z W, ZHANG S L, LIU R P, ZONG H T, JING Q, LI G, MA M Z, WANG W K. Electronic and optical properties of rock-salt aluminum nitride obtained from first principles [J]. J Phys: Condens Matter, 2007, 19: 425231.
[17] XIA Qing-lin, YI Jian-hong, LI Yan-feng, PENG Yuan-dong, Wang hong-zhong, ZHOU Chen-shang. First-principles investigations of the band structure and optical properties of γ-boron [J]. Solid State Commun, 2010, 150: 605-608.
[18] MONKHORST H J, PACK J D. Special points for Brillouin-zone integrations [J]. Phys Rev B, 1976, 13: 5188-5192.
[19] SUN Jian, WANG Hui-tian, HE Ju-long, TIAN Yong-jun. Ab initio investigations of optical properties of the high-pressure phases of ZnO [J]. Phys Rev B, 2005, 71(12): 125132.
[20] CHENG Y C, WU X L, ZHU J, XU L L, LI S H, CHU PAUL K. Optical properties of rocksalt and zinc blende AlN phases: First-principles calculations [J]. J Appl Phys, 2008, 103: 073707.
(Edited by HE Yun-bin)
Foundation item: Project(50474051) supported by the National Natural Science Foundation of China
Received date: 2010-12-16; Accepted date: 2011-04-06
Corresponding author: SHI Yi-min, Associate Professor; Tel: +86-15111157635; E-mail: hn_syb@126.com

