���ˮ����Dz�����������뷢չ
��Դ�ڿ����й���ɫ����ѧ��2017���6��
�������ߣ����� ���� �ܹ���
����ҳ�룺1289 - 1302
�ؼ��ʣ����ˮ�����ܣ����ⷴӦ���Dz�����
Key words��water electrolysis; hydrogen energy; hydrogen evolution reaction; non-Pt catalyst
ժ Ҫ��������Ϊһ����ࡢ���ܡ�������Ⱦ�ġ���ɫ��Դ��������Ϊ�Ǻ�ʯ��ʱ�������ԴΣ���ͻ���Σ����������Դ֮һ�����е��ˮ���⼼����Ӧ��Ч���ء�����Ⱦ���ص����Ϊ�о��ȵ㡣Ŀǰ����������õĴ�����Ȼ��Pt��������Ȼ��Pt�����١��۸����������ģ������������ʵ������ˣ���ν�һ��������з�Pt�������������ܻ����۸�Ч�����ʹ�����ʵ�ֵ��ˮ�����ģ�������Ĺؼ�����ϱ��������ڵ��ˮ������о������������������Pt����������о��½�չ���ص���������ɽ�������������ϵĽṹ�������ض��������ܵ�Ӱ�죬���Ե��ˮ����������ٵ���ս����չ�������չ����
Abstract: Hydrogen energy is a clean, high energy, environmentally friendly resource which is considered as one of the most promising candidates for replacing fossil fuels in the future. Electrochemically splitting water into hydrogen by renewable energy has attracted much attention due to its high efficiency, easy operation and no secondary pollution. Although the state-of-the-art catalyst for HER is a typical Pt-based catalyst, its scarcity and high cost prevent their widespread applications. Thus, it is highly desirable to exploit non-Pt HER catalysts with high activity, long-term stability and low cost. The recent advancements in the area of non-Pt HER catalysts with emphasis on introducing the exciting new research in the structure regulation of transition metal compounds and the understanding of the mechanisms of catalysts were reviewed. Meanwhile, the insights into the remaining challenges and research directions were proposed to shed light on future development of non-Pt HER catalysts.
DOI��10.19476/j.ysxb.1004.0609.2017.06.024
�� ������ �£��ܹ���
(���칤�̴�ѧ ������Դ��������װ�������������о�����
���뻷���²����������ص�ʵ���ң����� 400067)
ժ Ҫ��������Ϊһ����ࡢ���ܡ�������Ⱦ�ġ���ɫ��Դ��������Ϊ�Ǻ�ʯ��ʱ�������ԴΣ���ͻ���Σ����������Դ֮һ�����е��ˮ���⼼����Ӧ��Ч���ء�����Ⱦ���ص����Ϊ�о��ȵ㡣Ŀǰ����������õĴ�����Ȼ��Pt��������Ȼ��Pt�����١��۸����������ģ������������ʵ������ˣ���ν�һ��������з�Pt�������������ܻ����۸�Ч�����ʹ�����ʵ�ֵ��ˮ�����ģ�������Ĺؼ�����ϱ��������ڵ��ˮ������о������������������Pt����������о��½�չ���ص���������ɽ�������������ϵĽṹ�������ض��������ܵ�Ӱ�죬���Ե��ˮ����������ٵ���ս����չ�������չ����
�ؼ��ʣ����ˮ�����ܣ����ⷴӦ���Dz�����
���±�ţ�1004-0609(2017)-06-1289-13���� ��ͼ����ţ�TQ151.1��O643.36���� ���ױ�־�룺A
���Ź��õij������ٷ�չ���ҹ���Դ������������ú��ʯ�͡���Ȼ���ȴ�ͳ��ʯ��Դ�������ޣ��Ҵ����Ļ�����Ⱦ�������ء���ˣ�������Ч�������Ѻõ�������Դ�������������С�������Ϊһ����Դ�ḻ�����ܡ�������Ⱦ�ġ���ɫ��Դ��������Ϊ�Ǻ�ʯ��ʱ�������ԴΣ���ͻ���Σ����������Դ֮һ[1]������;���ж��֣����е��ˮ���⣬���ò��ɴ洢�Ŀ�������Դ(̫���ܡ����ܡ���ϫ�ܵ�)�����ˮ��������������������õIJ�Ʒ��ࡢ���ȸߣ������Խ����ܼ�Ӵ���Ϊ��ѧ�ܴ����ã��ر��ǽ���Ӧ�õ�ȼ�ϵ�������У����ô��������ܱ任Ϊ���ܣ������䷴Ӧ������û���κ���Ⱦ��ˮ��������ʹ������ת��Ч�ʴﵽ80%���ϣ����Ը�����ȼ��30%��ת��Ч�ʣ��Ӷ�ʵ������Դ�ɳ���ѭ������[2-3]��
���ܵ��ˮ���ⷴӦ(HER)���н��������ʷ���������о�����������Pt������������������HER����λ��Ȼ�ܴ�ʹ�õ��ˮ����Ч�ʲ��ߡ�Ȼ��Pt���������������١��۸���������ģ������������ʵ������ˣ������������о���������ν�һ��������з�Pt�������������ܻ����۸�Ч�����ʹ���[4-6]�����������������ˮ�����Pt�������о��½�չ�����ܽ���ˮ����������ٵ���ս����չ����
1 ������ⷴӦ����
����HER����[7-8]�����������о����о���Աͨ���ܽ����������Ϸ������ⷴӦ��ʵ�����ݺ��о�������������Ӧ�ķ�Ӧ���ۣ���HER������Ҫ������
1) �绯ѧ��Ӧ(Volmer��Ӧ)
���Խ����У�
H3O++e+M��M��Hads+H2O (1)
��/���Խ����У�
H2O+e+M��M��Hads+OH- (2)
ʽ�У�MΪ����������M��HadsΪ�������������γɵ�������ԭ�ӡ�
2) ת����Ӧ
��ʩ�ӵ��������£��������Ա������ɵ�M��Hads����������ʲ�ͬ�������ֲ�ͬ�ķ�ʽ����H2��
�ٵ绯ѧ�Ѹ�(Heyrovsky��Ӧ)�����������Բ���棬����һ��H3O+��M��Hadsλ���Ϸŵ�����H2��
���Խ����У�
M��Hads+H3O++e��M+H2+H2O (3)
��/���Խ����У�
M��Hads+H2O+e��M+H2+OH- (4)
�ڸ����Ѹ�(Tafel��Ӧ)�����������Բ���棬����M��Hads��������H2��
M��Hads+M��Hads��2M+H2 (5)
3) ��������
���������Բ�������ɵ�H2�ڵ缫�����ۼ�����̶������������缫���棬����Һ���ݳ���
nH2��H2�� (����) (6)
�κ�һ�ַ�Ӧ���̶���������绯ѧ����(Volmer��Ӧ)������һ���Ѹ�����(Heyrovsky��Ӧ��Tafel��Ӧ)�����ԣ���缫��Ӧ��������������ķ�Ӧ���̣�Volmer-Heyrovsky������Volmer-Tafel����[8]��Ȼ����������Ӧ�����У�������һ�����ٿز����о�ѧ�ߵĹ۵㲻һ�¡����У��ٻ��ŵ�������Ϊ�绯ѧ��Ӧ����(Volmer��Ӧ)�������绯ѧ�Ѹ���������Ϊ�绯ѧ�Ѹ�����Ϊ�ٿز���������������Ϊ�����Ѹ�����Ϊ�ٿز���
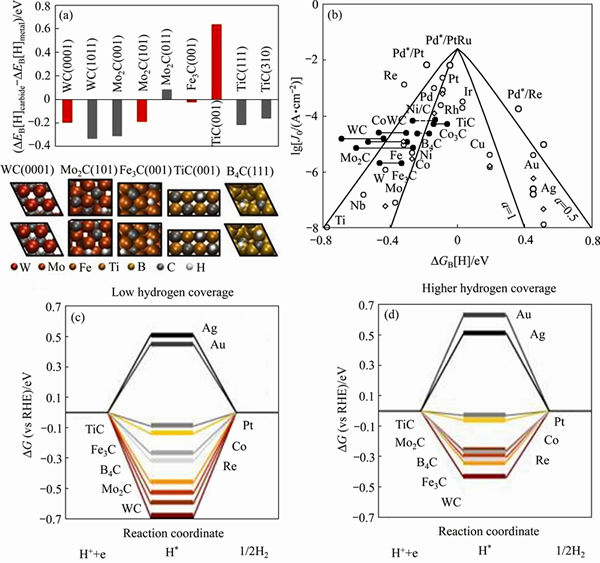
Ȼ�������������ֻ������У�HER�����Դ������������Ļ�����ԭ��Ϊ��Ӧ�м��壬��M��Hads��Ȼ���ٷ���M��Hads�������γ�H2���ɴ˿ɼ�������������HER�ĵ��������M��Hads�����������ء�����缫���У�ͨ�����ý��������ܶ�(J0)������������Ĵ������ͷ�Ӧ����ѧ�������о������������M��Hads����ܽ���ʱ������������H�ڴ��������Ѹ�����M��Hads����ܽ�ǿʱ����������H��������ʱ�����������Ѹ���绯ѧ�Ѹ�������Ҫ�˷��Ļ�ܽ������ӣ�����M��Hads�������γ�H2��������ʽ��ͣ��ܷ�Ӧ�����½������ԣ�ֻ����M��Hads�����ǿ������ʱH2������Ӧ�����ʲ��ܴﵽ���������缫������M��Hads�����֮��Ĺ�ϵ������س�Ϊ����ɽ��ЧӦ����
ͼ1��ʾΪ����[9-10]�����ܽ��HER���������ܶ���M��Hads��ǿ��֮��Ļ�ɽ��ϵͼ����ͼ1�п��������ع۲쵽����ͬ���������HER���������ܶȲ���ϴ����У������Pt���ڻ�ɽ���㸽�����Ի���H�������еļ��ܣ���������ڻ���H������/�Ѹ�����������á���ֵ��ע����ǣ�Pt��û�д��ڻ�ɽ���㣬���HER���Ի��������Ŀռ䡣������������M��Had����̫ǿ��̫�����ֲ��ڻ�ɽ�������࣬HER���������ܶȽ�С������Pt������������ͨ�����������뷴Ӧ�м����ڴ����������������ܶ��γɵĻ�ɽ�������Ǵ���Ӧ�д��ڵ�һ���ձ���ɣ������м���������е�������ǿ�Ⱥ��Ƕ�ʱ�����������ߵķ�Ӧ���ʣ�����Ϊ��Ƹ�Ч�����ṩ��ָ�����ݡ�

ͼ1 HER���������ܶ���M��H��ǿ֮��Ļ�ɽ��ϵ[9-10]
Fig. 1 Volcano plot of exchange current density in HER as a function of M��H bond strength[9-10]
2 �������Dz�����
��HER�����У�������һֱ���о��ߵ��о��ص㡣������������Ҫ��������ĵ��ӽṹ���������ӽṹ�����Ӽ��νṹ�ʹ����Ե���������ͼ1��ʾ����ε��ش����ı��滯ѧ���ʣ�ʹ�������H���ɵĻ�ѧ���������е�������ǿ�ȣ��������������H������/�Ѹ�������������Ч��������绯ѧ��Ӧ�����еļ�����������ǿ���������������ԡ�ͬʱ����ƴ����ṹ����Ч���ش����ı�����ò����ɢ�ԣ�ʹ������¶����Ļ���λ�����ϴ����������õĵ����Ժ���������������/���Һ����Ҳ��ͬ����Ҫ�ġ���ˣ�������Χ�����������棬��Pt��������õ���������չ�����潫��Ҫ�����Щ��������������۽��ܡ�
2.1 �Ͻ����
���ڶ��Pt�����У����ɽ���Ni����ԭ��������δ�ɶԵ�d���ӣ�����������Ӧ��������������ԭ��1s�������γ�Ni��H������ܹ���HER�ܺõĵ�����ԣ������������λ��Ȼ�ϸ�[11]������JAKSIC[12]����Brewer-Engel�ۼ�����Ԥ�⣬��d����������d�������Moϵ������d������С��d�������Niϵ�����γɺϽ�ʱ�������ڸĽ�����������H�Ľ���ܣ��⽫��HER����ЭͬЧӦ����ߴ����ԡ�RAJ[13]�о��˼��ֶ�Ԫ�Ͻ��HER���ԣ���С����ΪNi-Mo��Ni-Zn��Ni-Co��Ni-W��Ni-Fe��Ni-Cr������Ni-Mo�Ͻ𱻹㷺��Ϊ������ǰ��ʵ�ֲ�ҵ�����������Ĵ����ϡ��Դˣ�ZHANG��[14]���ôſؽ��似����Ni�����Ϲ����˾��ȵĶ�Ԫ�Ͻ�Ni-Mo���ߴ��һ��Ԫ�طֲ�����(��ͼ2)��Ni-Mo�Ͻ���нϸ�HER���Ե�ԭ����Ҫ��Դ��Ni-Mo�Ͻ�����ЭͬЧӦ�������Ǵ�ıȱ�����������������Ǵ��ڵĵ縺�Բ���ʹ������Ni(1.91)��Mo(2.61)ת�ƣ�����Mo��Χ�����˵��Ӹ������Ӷ��γ�Эͬ�����ã�������нϸߵĴ�����[17]��
Ȼ����Ni-Mo�Ͻ�Ʋ���Ӧ���ϴƲ�����帽����Ƿ�ѣ����ŷ�Ӧ�������У��Ʋ��е�MoԪ�ػ����ܽ⣬������HER����˥���Ͽ졣Ϊ�ˣ�һЩѧ���ڶ�Ԫ�Ͻ�Ļ����ϣ�ͨ�����������Ԫ�ؽ�һ���Ľ�Ni���Ͻ����ĵ��ӽṹ״̬����������Ĵֲڶȣ��Ӷ���ߴ�����HER���Ժ��ȶ��ԡ��߳ϻԵ�[15]���õ�������Ʊ��Ǿ�/����Ni-Mo-La�Ͻ�ϡ��Ԫ�صļ����������γɷǾ�̬Ni-Mo�Ͻ�ϸ����������С�Ʋ�Ӧ�䣬��HER����λ��Ni-Mo�Ͻ�Լ80 mV��WANG��[16]Ҳ���õ绯ѧ��������TiƬ�ϵ��ظߺ���NiMoZn��Ԫ�Ͻ�����Zn������ٽ��˷�Ӧ���ת�ƣ��������������ڴ�����������������Ͻ���Zn����ռ2%(Ħ������)ʱ��NiMoZn�������ŵ�HER���Ժ��ȶ��ԡ��������ӽ���Ԫ���⣬�ںϽ�������ǽ���Ԫ�أ�ͨ��������ƣ���ɻ��HER��������Ĵ���������NiMo�Ͻ�������ǽ��������γ�NiMoN��Ԫ�Ͻ�[17]������HER����������ߣ����������Ժͼ��Ե��Һ�ж����кܺõ��ȶ��ԣ�ZHANG��[18]���õ��������ֽ̼���Ƶ�3D�ṹ��CoMoSx����������䷴Ӧ��ʼ����λ����100 mV����ʹ�����ܶ�Ϊ100 mA/cm2ʱҲֻ��180 mV�Ĺ���λ���ɴ˿ɼ����������ӵĵ�����Ԫ���ǽ������Ƿǽ��������Ƕ����Խ�һ���Ľ�Ni���Ͻ����ĵ��ӽṹ״̬��ṹ��ò���Ӷ���ǿHER���ܡ�
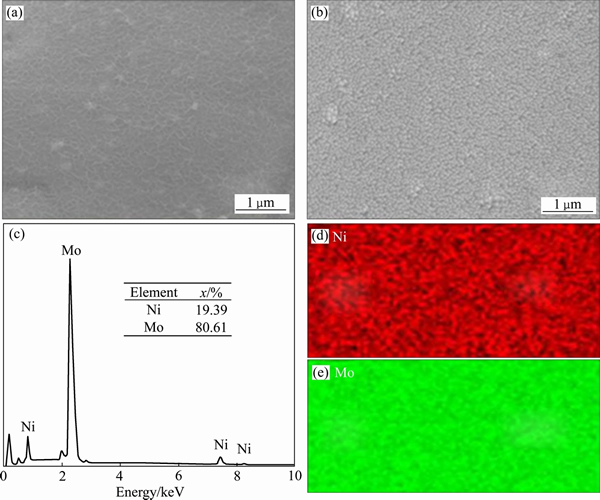
ͼ2 ��ĭNi����ĭNi-Mo/Ni��FE-SEM���Լ���ĭNi-Mo/Ni��EDS������Ԫ�طֲ�ͼ[14]
Fig. 2 FE-SEM images of Ni foam(a), Ni-Mo/Ni foam(b) and EDS results along with elemental mapping of Ni-Mo/Ni((c)-(e))[14]
2.2 ���ϴ���
ͨ���Ͻ���ߴ����ıȱ�������ǹ������������Ѿ߱��ϺõĴ����Ժ��ȶ��ԣ�����������ȣ����ǵĹ��д�������Ȼ��С������������Ϳ���·��ʴ�����ϲ�����ڹ�������ԣ������ִ�����Դϡ�١��۸������ڹ�ҵ�������д��ģʹ�á����ѡ�������е��κ�һ����Ϊ������ϣ��ܻ���ڲ�������⡣��ͨ�������IJ�����ƣ�����һ�������и��ϣ�ʹ��ּ�����ܵõ����ƻ������ﳤ�̣ܶ�����������ɴ����ϵ���Ҫ��������ȱʧ�����ܻ�ø��õ�Ч�����ҳɱ�Ҳ���������½�[19]��SOLMAZ��[20]���õ绯ѧ���������������Ag�� Pd��Pt���������NiCoZn�缫�ϣ��Ӷ���������������д����ԣ����������ܶ�������λ���ͣ��ڵ����ܶ�Ϊ10 mA/cm2�³�����Ӧ120 h������û�з����κα仯�������[21]���ú�������ϵ�����Ʊ�(Ni-W-P)-TiO2���ϵ缫����100 mA/cm2�������Ni-W-P ������176 mV��������Ϊ���ϵ缫HER����������ߵ�ԭ���Ƿ�Ӧ�����ĸı䣬TiO2����������Ni-W-P �Ͻ�������Եĵ���ЭͬЧӦ��VAZQUEZ-GOMEZ��[22]���ø��ϵ����������˾��ж�ṹ�� Ni+RuO2�������������RuO2�����벻�����������HER���ԣ�����Ҫ������ǿ�˿���������Ϳ���·��ʴ����������ڼ�����Һ�о��кܺõĵ�����Ժ��ȶ��ԡ�
�ܴ�������XIONG��[23]���øߵ�����������������������Ϊ��ģ�塱�������Ƶ����ʡ��������Ķ�������������ıȱ�����������õ�����������и���������Ե������ֳ�����������У�ʹRu������Ni�ԳɶԵ���ʽ���ڣ�Ni�Ե��ʺ���������ʽ���ڣ��ٽ�����H��ת�ƣ��Ӷ�����ЭͬЧӦ����������ʼ��λ������Pt��������������Ե��ڴ�RuO2��p-Ni��ƽ��Ni��(��ͼ3)��
�ɴ˱�����RuO2/p-Ni��HER���Կ���Pt����������������������λ�����RuO2/p-Ni��HER����Խ��Խ�ã���������Pt���������ø��ϴ�����������ʼ��λ��Pt������������������γ�ЭͬЧӦ����������д��������ڵ�һ���������ڴ�����ܶ�����ʾ�����õ����������鹦�ڸø��ϴ������и���Ļ���λ��¶����������С����⣬ZHANG��[24]������ˮ�ȳɷ�Ҳ�ɹ�������������ԭλ�Ʊ�����������������ɢ��RuO2-NiO���װ����нṹ�����к��������٣���HER���Ըߣ���ʹ��500 mA/cm2�Ĵ�����ܶ��½��г�����Ӧ100 h������Ȼ��ʾ���ܺõ�HER���Ժ����õ��ȶ��ԡ�

ͼ3 Ni��p-Ni��RuO2��Pt��RuO2/p-Ni������6 mol/L NaOH���Һ�е����⼫������[23]
Fig. 3 LSV of Ni, p-Ni, RuO2, Pt, and RuO2/p-Ni catalysts in a 6 mol/L NaOH electrolyte[23]
2.3 ���ɽ�������
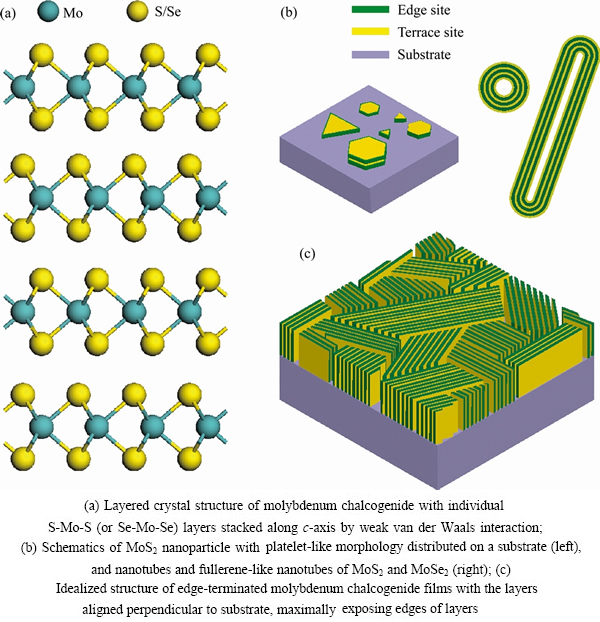
�����������ڹ��ɽ�������ر���������о��dz����˹�ע��Ȼ�������о���Ϊ����״����û��HER���ԡ�ֱ��2005��ű�HINNEMANN��[25]�߸�������ͨ�����ۼ��㷢�֣�����Ĵ�����λ��λ�ڲ�״�ṹ�ı�Ե��������Hԭ�ӵĽ������Pt�������Ϊ������Ϊһ����Ч����������ṩ�����ۻ�����
�Դˣ�����ͨ��ʵ�飬�����⸺����ʯī��Ӧ����HER��Ӧ��֤ʵ�����׳߶ȵ��������һ����HER���ԡ������о��ѱ�������ǿ�����������ܵ�;����Ҫ��ͨ��������Ե�ṹ�Ļ���λ��(����������ԭ��)����ߴ��������Եķ�ʽʵ��[26-31]��KIBSGAARD��[26]����˫��������SiO2Ϊģ���Ƶý��MoS2�缫�����ֶ��صĽṹʹ����ı�Ե�õ����ȱ�¶(��ͼ4)���Ӷ���������������Ļ���λ����Ŀ��KONG��[27]���������˼��Ӧ�õ����������������У���ͼ5��ʾ�����û�ѧ�������������ֱ�ڻ���(Mo/W)S2��(Mo/W)Se2�����ӱ�¶���Ա�Եλ����Ŀ�������������Ҳ�õ����������ӡ�Ȼ����������������ǵ��͵ļ�Ӵ�϶�뵼�壬��ϲ�ĵ��Ӵ���������һ���̶������������ķ�չ��
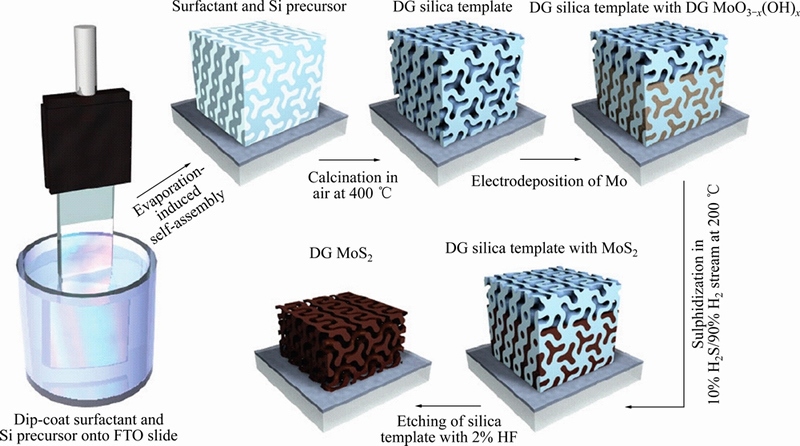
ͼ4 �������ȱ�¶���Ա�Եλ���˫��������MoS2������ʾ��ͼ[26]
Fig. 4 Schematic diagram of synthesis procedure and structural model for mesoporous MoS2 with double-gyroid morphology[26]
ͼ5 ��ֱ���е��⻯�������ṹʾ��ͼ[27]
Fig. 5 Nanostructures of layered MoS2 and MoSe2[27]
������������LUKOWSKI��[28]ʹ��������﮶�������ʯī�����ϵ�MoS2���в����룬�õ��˾��н������Ե�1T��MoS2��������Ƭ����ͬ�������£����õ�1T��MoS2��ʾ����������������Ժ��ڳ���2H��MoS2����1T��MoS2��һ������̬�ṹ�����ŷ�Ӧ�������У�������ת��Ϊ�����Խϲ��2H�ͣ��������Ҳ��˥�ˡ�STASZAK-JIRKOVSKY��[29]�Ʊ�������CoSx�������ܾ��н϶���λ�����͵���ԭ�ӣ�������Խϸߣ�ͬ��Ҳ�����ȶ��Խϲ�������TANG��[30]��̼���Ϲ�������3D�ṹ��NiS2�������У������Խ����У������ܶ�Ϊ10 mA/cm2ʱ�������λֻ��243 mV�����ƶ�б��Ϊ69 mV/(��)�������DENG��[31]�Ե��������õ�ʯīϩΪ���壬����ˮ�ȷ��ϳ��˳�������������Ƭ��ֱ��ʯīϩ(MoS2��RGO)��Ϊһ����Ч����������������ʾ����������������Ƭ���ȵش�ֱ������ʯīϩ�ϣ��缫�ĵ�����������ǿ�����ұ�¶�����ӷḻ�ı�Եλ�㣬������ֳ��ߵĴ����Ժ����õ��ȶ��ԡ�
���⣬��������������ԭ��Ҳ����ߴ����Ĵ����Ժ��ȶ��ԡ�MERKI��[32]���֣�Fe��Co��Ni���ε�����������нϴ���������ŵ�HER���ԡ���ϵ�д������Ե������Ҫ�����ںϽ���ǿ�˴����Ĺ��д����ԣ�����˴����ķ�ɢ�ȣ��ɴ������˴����ȱ�����ͷ�Ӧ����λ��REN��[33]��ͬ��Ԫ���н����Ը��õ�Se���뵽MoS2�У��ڹ���λΪ400 mVʱ��������ܶȴﵽ42.7 mA/cm2����δ����MoS2��3����MIAO��[34]�Ʊ���Ni-MoS2��Ni�IJ��Ӹı���MoS2��������ò���γ��˴�����ȱ��λ�ã���С�˴����Ķѻ��;ۼ��̶ȣ������HER���Ժ��ȶ��Եõ�������ߡ�
2.4 ���ɽ���̼����
���ɽ���̼����ӵ�кܸߵ�Ӳ�ȡ��Ϻõ��ȶ��Ժͽ�ǿ����ʴ�ԣ���һ�����͵Ĺ��ܲ��ϣ��ڸ������¡���ĥ�����ͻ�ѧ��ʴ�Ļ�е�����ѻ�ù㷺ʹ�á����������о�ѧ�߷��֣��������ʾ���һ���Ĵ����ԣ���������⡢��⡢���������͵���ȷ�Ӧ�б��ֳ������ڹ���������ʣ���˱���Ϊ����Pt�����������У�̼������к�ǿ�Ľ�����������������Ŀ����ж�������������Ŀ������Ҫԭ������̼ԭ�ӽ����׳�����4�����ӵ����ɽ���Ԫ�ص�d���ӣ�����Ԫ�����ڱ��е�����4��λ��Ԫ����ͬ������Pt����ͬ����ʱ����ĵ������Ը��ӽ���Pt��������������������Pt������Ĵ�����[35]��MICHALSKY��[36]��һϵ�й��ɽ���̼���������ʵ�������ۼ����о�(��ͼ6)���봿������ȣ��γɵ�̼��������и��õĴ����ԣ�������Ϊ����̼������γɵ��½���ԭ��������H֮��Ľ�������������������ܽ�ġ���ɽЧӦ������ֻ����M��Hads�����ǿ������ʱ����������Ӧ�����ʲ��ܴﵽ����ǽ���̼������ӻ��γɵ�̼���ﵼ�½���ԭ��������H֮��Ľ������������ʹHER�Ĵ����Ե�����ߡ����ڶ���ɽ���̼�����У�̼����(Mo2C)��̼����(WC)�Լ�̼����(TaN)��Ϊ����HER�����ԽϸߵĴ����ܵ��˽϶���о�[37-43]��
WEIDMAN��[37]���ֵڢ�����Ĺ��ɽ����γɵ�̼�����ڽϿ���pH(0~14)��Χ�ھ����нϺõĻ�ѧ�ȶ��ԡ�WC��W2C��Mo2C�����ⷴӦ��缫������������ʼ��λ�ȽϽ��������Mo2C��pHΪ0ʱ���к�С���������λ(~70 mV)��WC���и��õĿ�������������õ��ȶ��ԣ���ʹ���Һ��pHС��2.5������Ȼ����W2C��Mo2C����˿�����Ϊһ�����Խ����еĴ�����������塣VRUBEL��[38]ֱ�ӽ���ҵMo2CӦ���ڵ�����ⷴӦ�У�������֣�Mo2C�����Ժͼ�����ϵ�ж����ֳ���һ����������ԣ�����������һ��ֻ�в�������Ż���С�Ȼ���������������λ�ϴ�����Pt������������Ҫԭ��������ҵMo2C�Ĵ��Ȳ��ߣ��ߴ�ϴ��Աȱ����С��������[39]��������Ϊǰ���壬ͨ��ˮ������װ�ϳ���������Ƭ���ٽ��仹ԭ̼�����̼��������Ƭ���ڵ����Ӧ�о����벬���ƵĴ����ܡ��Դˣ�CHEN��[40]������笠��ص�̿�ں�̼�����ϣ��ڶ��������и��±��գ�����������Ϊ̼Դֱ��̼�������Σ��Ӷ�������ṹ��Mo2C/XC-72��Mo2C/CNT������Mo2C���ȷ�ɢ�������ϣ�������ΧΪ7~15 nm������ҵMo2C��Աȣ������ʹ�����չ�ֳ����õ���������ԡ������������������������ΪMo2Cê����̼�����ϣ�һ��������˴����ž�����ʹ����λ��ֱ�¶������ͬʱҲ�����˵��Ӵ���·�����Ӷ�����˴����ԣ���һ���棬����ê��ЧӦ�յ���ɴ���ת��̼��ʹ���d���������ƣ������˻���H�������ܣ��������ⷴӦ�������С�LIAO��[41]Ϊ�����Mo2C����������ԣ����ñ���������Ϊ̼Դ��������什��оۺϣ��γ�MoOx/���������ߣ����ڶ��������и��±��յõ������Mo2C�����ߴ���������һά�����߽ṹ�����˵���ת��·�����ḻ�Ŀṹ��֤���Һ�ܹ�������ɢ������λ�ϣ������ʾ���Ϻõ���������ԡ�WAN��[42]ͨ������ǰ����ı����Ƶò�ͬFe��������Mo2C������Fe���������Mo2C��̬�����ʯī��̼�������ӣ����ڱ���������Fe2(MoO4)3��HER������ߡ������о�����̼���������HER�����Թ�����̼ԭ������ԭ�ӵ��ӻ�������ԭ�ӵĻ�ѧ���ʷ����仯��Ȼ�������о���δ��ȫڹ�����е�ԭ�����ȱ����ص��������ݣ������˽�һ���Ż��������������HER���ܣ������Ҫ�����о�̼��������Ĵ�������Ϊ�����HER�����ṩָ�����ݡ�
ͼ6 ��ͬ���ɽ���̼�����������Ӧ��������ԭ�������ܲ�ֵ(a)����ɽ��ϵͼ(b)����ͬ��ԭ�Ӹ������¶��ֵ��������ԭ������������(c), (d)[36]
Fig. 6 Difference in H adsorption energies on various metal carbide surfaces relative to their stepped parent metal surface(a); volcano plot for HER on metal surface(b); free energy diagrams for electrochemical reduction of H+ at equilibrium potential and under standard conditions(c), (d)[36]
�Դˣ�XIONG��[43]ͨ��ʵ�������ۼ������̽����Ni����̼��������ı�����ӽṹ�仯��ϵ��HER�����ܵ�Ӱ�졣����һ�ּɿص�ˮ�Ⱥϳɷ�ֱ������ά�ṹ����ĭ������������һ������ǰ���壬��ͨ��������̼����ת��Ϊһά�ṹ��NiMo2C���װ����ϴ�������ͼ7��ʾ������ֱ�����������װ��ṹ���Թ�����Ч����ά��������ĵ��ӡ���Ӧ��/������ͨ����������ֵ��͵��ѷ��ʹ��ճ�ϼ����缫�Ļ���λ������ȵر�¶�ڹ�/Һ�����ϣ���֤�������и��õ���������Ժ��ȶ��ԡ�Ϊ�˷���Ni����̼��������ı�����ӽṹ�仯��ϵ��HER�����ܵ�Ӱ�죬�������ۼ���ģ���˴�Mo2C��NiMo2C����ļ��Ρ����ӹ��ͺ�����H��ǿ�ȡ���ͼ7�п�������Mo2C (001)�����ɷֲ����ȣ���ɫ��ͬ����Ni���ӵ�Mo2C�У�NiMo2C (001)����Mo�ĵ�ɷֲ���ʼ�����仯������Ni���Դ��ٽ���Mo��õ��ӣ��Ӷ���ɱ���Mo�ĵ�ɷֲ������ȡ�NiԽ����Mo��Խ���״�Mo�����õ��ӣ��Ӷ�ʹMo��������������ɣ���Ni���������ĸ���ɡ����������Ȼ�����������ˮ�н����H������������Ϊ��ȷ��H��Mo2C(001)��NiMo2C (001)������������Գ̶ȣ���һ��������H���������á��������������Ni�����ı���Mo2C����ĵ������ʣ���ʹNiMo2C������H��ǿ�������½�������H����˹������������0����ˣ�NiMo2C�������е�H�����ܣ������ڴӴ��������Ѹ�����H2���뿪�������棬ʹ����Hռ�ݵĻ���λ��ʱ�ͷų����������뷴Ӧ���Ӷ�ʹ���ⷴӦ���Գ������С������ۼ�������ʵ����һ�¡�
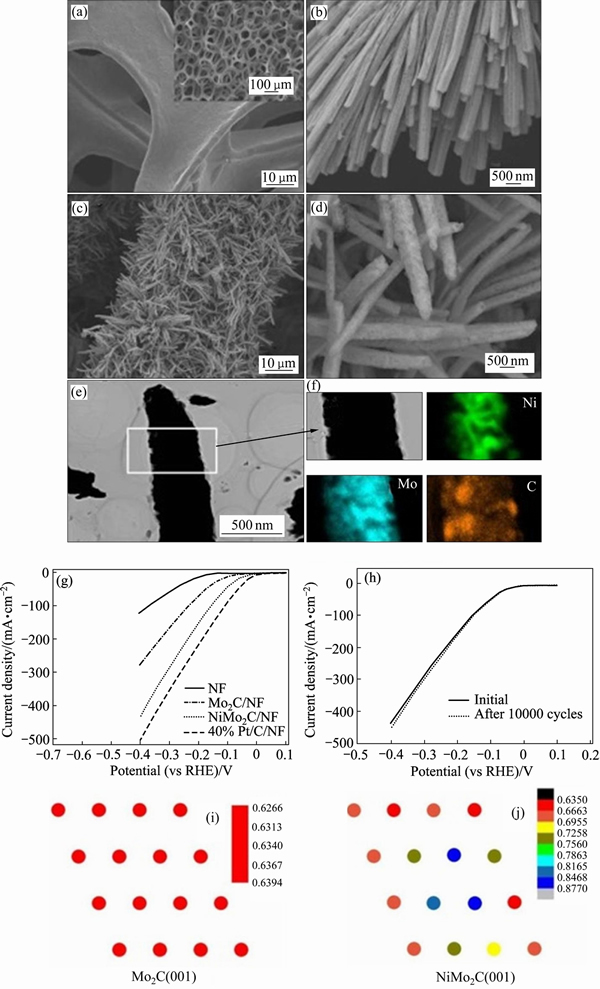
ͼ7 ������SEM/TEM��ӦHER�������ߺ�Mo2C (001)��NiMo2C (001)����Bader��ɷֲ����[43]
Fig. 7 SEM/TEM images of catalysts and corresponding HER activity ((a)-(h)) and distribution of Bader charge of Mo2C (001) and NiMo2C (001) ((i), (j))[43]
2.5 ���ɽ�������
���ɽ�������Ҳ��һ��dz���Ҫ�������ܲ��ϣ���������ɽ���̼�������Ƶĵ����ԡ������ԡ���Ӳ�ȡ���ǿ�Ⱥ��ȶ��Ե��ŵ㣬�ѱ��㷺Ӧ���ڴ�����Դ��﮵�ء���ѧ������������������[44-45]�����ɽ�������ĺϳɣ����������׳߶��¿�������ò�ͽṹ����Ϊ���Ϻϳ������һ���ȵ㡣���������о���Ա�����˲�ͬ�ĺϳɷ����Ʊ��������ײ� ��[46-48]��MUTHUSWAMY��[47]ѡ�����Һ�������������������(TOP)Ϊ�Դ�������ֹ��ɽ���������ת��Ϊ����ڴˣ��ص�����������ֵ��͵�����ṹ��MoP�����������ṹ[49]��Moԭ����Χ����6��Pԭ�ӣ�MoP��(001)����5�����Ӳ���ɣ�����������һ�����ɳ�״̬���ڣ����������Խ���״̬���ڣ��Ӷ�ȷ������Ľṹ�ȶ���Ni2P���ǵ��͵�Fe2P��������ṹ[50]��Pԭ�ӵIJ���ʹ��Niԭ��֮��ļ���ø�����Ȼ��Niԭ�Ӻ�Pԭ�ӵļ����϶̣�����γ���ԭ���ܼ��ľ���ṹ���Ӷ�ʹ��¶�ڱ����Pԭ����Niԭ��֮������˼���ЧӦ����ɱ�������������Ӻ�����ӵĽ������ģ�ΪNi2P��Ϊһ�ָ�Ч��������ṩ�˻������ġ�
POPCZUN��[51]����һ�ּ��ܼ��ȷ��ϳ����׳߶ȵ��������������кܺõ�HER�����ԣ����ƶ�б��Ϊ46 mV/decade�����������ܶ�Ϊ3.3��10-5 A/cm2���ڵ����ܶ�Ϊ20 mA/cm2ʱ������λֻ��130 mV���ڴ˻����ϣ�POPCZUN��[52]�Ʊ������ܴ�����20 mA/cm2�ĵ����ܶ��¹���λ���͵�85 mV��ͬʱ�������������ʾ���ϸߵĵ绯ѧ�ȶ��ԣ�ʹ���Ϊһ�����Ӧ��ǰ����HER������ֵ��ע����ǣ����ܹ��ɽ����������ײ��ϵĺϳ��Ѿ����˳���Ľ��������������临�ӵĺϳ�·����ʹ���л������������ģ����������ˣ�Ѱ�������Ч�ķ���ʵ�־��ж�����ṹ���ɽ�������ĺϳ����Ǹ��о������е�һ���������ս��
Ϊ�˼������Ʊ��������ж����ʺ�Σ�������ʹ�ã�SUN��[53-55]ѡ�����������Ϊ��Դ�����õ��¹�������Ӧ�ϳ���һϵ�е�HER�����������ˮ�ȷ��ϳ�FeOOH NA/Ti��Ni(OH)x/Tiǰ���壬��������������������������300 ���±���1 h���Ƶ�HER���������FeP/Ti��Ni2P/Ti��������̼��(CC)������Co(OH)F���������У��ٵ������õ���֧�Ŷ��CoP����������(CoP/CC)��ͼ8��ʾ��������άCoP/CC�ṹ��������ֱ�������绯ѧ������������0~14 pHֵ��Χ����ʾ������Ĵ����ԣ���Ŀǰ�������ŵ�Pt/C���Բ����С������8000 s������⣬��Ȼ���ֺܺõ��ȶ��ԡ�
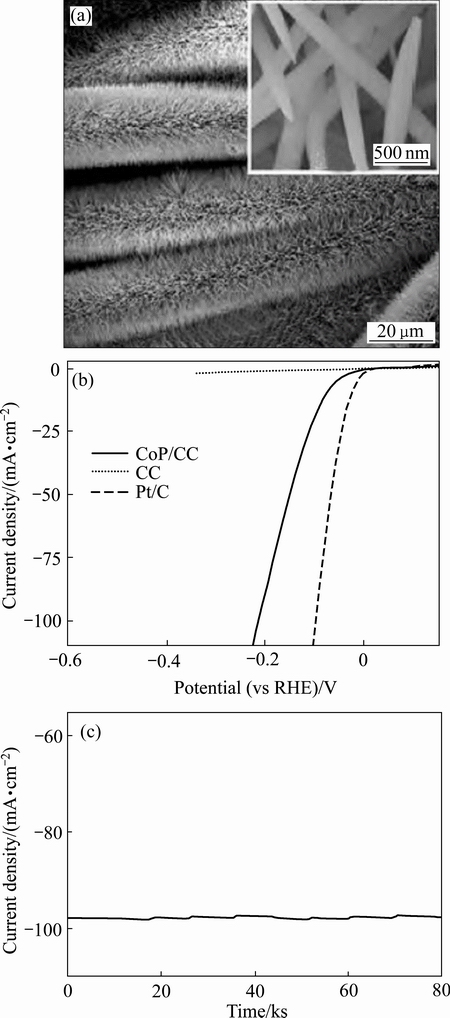
ͼ8 CoP/CC��SEMͼ��CoP/CC��CC��Pt/C ��0.5 mol/L H2SO4��Һ�е�HER���������Լ��ڹ���λΪ200 mV�²��Ե��ȶ�������[53]
Fig. 8 SEM images of CoP/CC(a), polarization curves of CoP/CC, blank CC and Pt/C in 0.5 mol/L H2SO4(b) and time-dependent current density curve for CoP/CC under static overpotential of 200 mV for 80000 s(c)[53]
XIAO��[56]�ܴ����������ù����Ƚԭ������������李���������������������ܽ����±��գ�ͨ������H2��ԭ���γ�Mo3P��MoP������������������Ժͼ��Խ����о����нϺõ�HER���ԣ����Ҳ�ͬ�����̶ȵ�����HER���ܲ������ԡ�ͨ�����ۼ���������֣��ô����Ļ���λ��Pԭ�ӣ�������������а��ݵĽ�ɫ�����������е�Sԭ�ӣ���һ����H���Ƕ��£��Ի���H�����������У����нӽ���0�ļ���˹�����ܣ������ʾ�����õ�HER���ܡ�KIBSGAARD��[57]��S����MoP�ı��棬����S����MoP����P�ĵ��ӽṹ����Ч���ƴ������汻�������Ӷ�ʹ������HER����������ߣ��ڵ����ܶȴﵽ10 mA/cm2ʱ�������λֻ��86 mV����ˣ�ͨ����ͬ�������ع��ɽ����������P�ĵ������ʿɻ�÷ḻ�Ļ���λ�㣬Ϊ�����ʹ�������λ���̽���ṩ���µ�˼·��
3 ����
���������������������Ǵ�ʵ������۽ǶȶԵ�������������Ƽ����ܸ��ƻ��Ƶ����ⷽ����ȡ���˾������������DFT�����ṩ�����ඨ�ԺͰ붨���Ľ�������ǣ�Ŀǰ��Ȼ������ȫ�������ۼ��������缫��Ӧ�����Ͷ���ѧ��ȷ��������⣬��ҺpH����缫��Ӧ�����Ͷ���ѧ��Ӱ�켰�䱾�ʡ����ͷǹ��������(���������̼�������������������)��HER�Ĵ������ؼ�����ָ�����ؼ����湤����Ʒ������ʶ��Ȼ���ںܴ�IJ��㡣��ˣ��ڵ�ǰ�Է�Pt������������ʶ��ʵ���о��Ļ���֮�ϣ����д��ڷ�չ���������ȵ�ʵ�鼼�����ڴ����ϵĽṹ�뷴Ӧ�����ɿؾ�ȷ��������»��ԭλ�ķ���ˮƽ�ϵ�֤�ݣ���չ��Ϊ�ӽ��绯ѧ��ϵʵ�����ε�����ģ�ͣ���ԭ�ӡ�����ˮƽ��ȷ���������ͷDz������Ļ���λ�㣬̽����Pt��������Ļ�����ԭ����ɡ����ӽṹ��������ò�Ĺ�Ч��ϵ������������Ч����λ�ܶȵļ����������ṹ���ŵ����ͷ�Pt�����������ߴ����ڷ�Ӧ�����е��ȶ��ԣ���δ�����ˮ��������о���չ����Ҫ����
REFERENCES
[1] ANDREWS J, SHABANI B. Re-envisioning the role of hydrogen in a sustainable energy economy[J]. International Journal of Hydrogen Energy, 2012, 37: 1184-1203.
[2] ������, ���ЭZ, ������, �� ��, �� ѩ. ��״˫���������������ڴ�ˮ�������о���չ[J]. ����ѧ��, 2016, 67(1): 54-72.
WANG Rui-rui, ZHAO You-jing, SHAO Ming-fei, XIANG Xu, DUAN Xue. Recent progresses in water oxidation over layered double hydroxide catalysts[J]. CIESC Journal, 2016, 67(1): 54-72.
[3] HOLLADAY J D, HU J, KING D L, WANG Y. An overview of hydrogen production technologies[J]. Catalysis Today, 2009, 139: 244-260.
[4] URSUA A, GANDIA L M, SANCHIS P. Hydrogen production from water electrolysis: Current status and future trends[J]. Proceedings of the IEEE, 2012, 100: 410-426.
[5] PLETCHER D, LI X. Prospects for alkaline zero gap water electrolysers for hydrogen production[J]. International Journal of Hydrogen Energy, 2011, 36: 15089-15104.
[6] ť����, �� ��, ������, ������, �� ��, �Ų���, ����ʤ. ����̼��ά�ı�����Զ�ˮ������ⷴӦ�����Ե�Ӱ��[J]. ��ѧѧ��, 2015, 73: 729-734.
NIU Dong-fang, DING Yong, MA Zhi-xing, WANG Ming-hui, LIU Zhou, ZHANG Bo-wen, ZHANG Xin-sheng. Effects of surface modification of carbon nanofibers on their electrocatalytic activity for hydrogen evolution reaction of water electrolysis[J]. Acta Chimica Sinica, 2015, 73: 729-734.
[7] BOCKRIS J O��M, POTTER E C. The mechanism of the cathodic hydrogen evolution reaction[J]. Journal of the Electrochemical Society, 1952, 99: 169-186.
[8] SHENG W C, GASTEIGER H A, SHAO-HOM Y. Hydrogen oxidation and evolution reaction kinetics on platinum: acid vs alkaline electrolytes[J]. Journal of the Electrochemical Society, 2010, 157: 1529-1536.
[9] CONWAY B, JERKIEWICZ G. Relation of energies and coverages of underpotential and overpotential deposited H at Pt and other metals to the ��volcano curve�� for cathodic H2 evolution kinetics[J]. Electrochimica Acta, 2000, 45: 4075-4083.
[10] TRASATTI S. Work function, electronegativity, and electrochemical behaviour of metals: ��. Electrolytic hydrogen evolution in acid solutions[J]. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry, 1972, 39: 163-184.
[11] CORREIA A N, MACHADO S A S, AVACA L A. Studies of the hydrogen evolution reaction on smooth Co and electrodeposited Ni-Co ultramicro electrodes[J]. Electrochemistry Communications, 1999, 1: 600-604.
[12] JAKSIC M. Electrocatalysis of hydrogen evolution in the light of the Brewer-Engel theory for bonding in metals and intermetallic phases[J]. Electrochimica Acta, 1984, 29: 1539-1550.
[13] RAJ I A. Nickel-based, binary-composite electrocatalysts for the cathodes in the energy-efficient industrial production of hydrogen from alkaline-water electrolytic cells[J]. Journal of Materials Science, 1993, 28: 4375-4382.
[14] ZHANG L, XIONG K, NIE Y, WANG X X, LIAO J H, WEI Z D. Sputtering nickel-molybdenum nanorods as an excellent hydrogen evolution reaction catalyst[J]. Journal of Power Sources, 2015, 297: 413-418.
[15] �߳ϻ�, �� ��. ������Ǿ�/����Ni-Mo-La�Ͻ�缫����������[J]. �й���ɫ����ѧ��, 2011, 21(11): 2819-2824.
GAO Cheng-hui, LI Ning. Hydrogen evolution reaction activity of electrodeposited amorphous/nanocrystalline Ni-Mo-La alloy electrode[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(11): 2819-2824.
[16] WANG X, SU R, ASLAN H, KIBSGAARD J, WENDT S, MENG L, DONG M, HUANG Y, BESENBACHER F. Tweaking the composition of NiMoZn alloy electrocatalyst for enhanced hydrogen evolution reaction performance[J]. Nano Energy, 2015, 12: 9-18.
[17] WANG T, WANG X, LIU Y, ZHENG J, LI X. A highly efficient and stable biphasic nanocrystalline Ni-Mo-N catalyst for hydrogen evolution in both acidic and alkaline electrolytes[J]. Nano Energy, 2016, 22: 111-119.
[18] ZHANG N, MA W, JIA F, NIU L. Controlled electrodeposition of CoMoSx on carbon cloth: A 3D cathode for highly-efficient electrocatalytic hydrogen evolution[J]. International Journal of Hydrogen Energy, 2016, 41: 3811-3819.
[19] ��ɭ��, ��Ǯ��. �����Ni-S/LaNi5����ϵ缫�ĵ����������[J].�й���ɫ����ѧ����2013,23(8): 2221-2228.
WANG Sen-lin, DUAN Qian-hua. Electrocatalytic hydrogen evolution characters of electrodeposited Ni-S/LaNi5 porous composite electrode [J]. The Chinese Journal of Nonferrous Metals, 2013, 23(8): 2221-2228.
[20] SOLMAZ R, KARDAS G. Fabrication and characterization of NiCoZn-M (M: Ag, Pd and Pt) electrocatalysts as cathode materials for electrochemical hydrogen production[J]. International Journal of Hydrogen Energy, 2011, 36: 12079-12087.
[21] ���, ������, �� ��. ������Ʊ�(Ni-W-P)-TiO2�����ϵ缫�Ĵ���������[J]. �й���ɫ����ѧ��, 2010, 20(4): 712-717.
LI Ai-chang, LUO Peng-fei, LIU Ying. Hydrogen evolution properties of (Ni-W-P)-TiO2 composite coating as electrode materials prepared by electrolytic co-deposition[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(4): 712-717.
[22] VAZQUEZ-GOMEZ L, CATTARIN S, GUERRIERO P, MUSIANI M. Preparation and electrochemical characterization of Ni+RuO2 composite cathodes of large effective area[J]. Electrochimica Acta, 2007, 52(28): 8055-8063.
[23] XIONG K, LI L, DENG Z H, XIA M R, CHEN S G, TAN S Y, PENG X J, DUAN C Y, WEI Z D. RuO2 loaded into porous Ni as a synergistic catalyst for hydrogen production[J]. RSC Advances, 2014, 4: 20521-20526.
[24] ZHANG L, XIONG K, CHEN S G, LI L, DENG Z H, WEI Z D. In situ growth of ruthenium oxide-nickel oxide nanorod arrays on nickel foam as a binder-free integrated cathode for hydrogen evolution[J]. Journal of Power Sources, 2015, 274: 114-120.
[25] HINNEMANN B, MOSES P G, BONDE J, JORGENSEN K P, NIELSEN J H, HORCH S, CHORKENDORFF I, NORSKOV J K. Biomimetic hydrogen evolution: MoS2 nanoparticles as catalyst for hydrogen evolution[J]. Journal of The American Chemical Society, 2005, 127(15): 5308-5309.
[26] KIBSGAARD J, CHEN Z, REINECKE B N, JARAMILLO T F. Engineering the surface structure of MoS2 to preferentially expose active edge sites for electrocatalysis[J]. Nature Materials, 2012, 11: 963-969.
[27] KONG D, WANG H, CHA J J, PASTA M, KOSKI K J, YAO J, CUI Y. Synthesis of MoS2 and MoSe2 films with vertically aligned layers[J]. Nano Letters, 2013, 13(3): 1341-1347.
[28] LUKOWSKI M A, DANIEL A S, MENG F, FORTICAUX A, LI L, JIN S. Enhanced hydrogen evolution catalysis from chemically exfoliated metallic MoS2 nanosheets[J]. Journal of The American Chemical Society, 2013, 135: 10274-10277.
[29] STASZAK-JIRKOVSKY J, MALLIAKAS C D, LOPES P P, DANILOVIC N, KOTA S S, CHANG K C, GENORIO B, STRMCNIK D, STAMENKOVIC V R, KANATZIDIS M G, MARKOVIC N M. Design of active and stable Co-Mo-Sx chalcogens as pH-universal catalysts for the hydrogen evolution reaction[J]. Nature Materials, 2016, 15: 197-203.
[30] TANG C, PU Z, LIU Q, ASIN A M, SUN X. NiS2 nanosheets array grown on carbon cloth as an efficient 3D hydrogen evolution cathode[J]. Electrochimica Acta, 2015, 153: 508-514.
[31] DENG Z H, LI L, DING W, XIONG K, WEI Z D. Synthesized ultrathin MoS2 nanosheets perpendicular to graphene for catalysis of hydrogen evolution reaction[J]. Chemical Communications, 2015, 51: 1893-1896.
[32] MERKI D, VRUBEL H, ROVELLI L, FIERRO S, HU X. Fe, Co, and Ni ions promote the catalytic activity of amorphous molybdenum sulfide films for hydrogen evolution[J]. Chemical Science, 2012, 3: 2515-2525.
[33] REN X P, MA Q, FAN H B, PANG L, ZHANG Y, YAO Y, REN X, LIU S. A Se-doped MoS2 nanosheet for improved hydrogen evolution reaction[J]. Chemical Communications, 2015, 51: 15997-16000.
[34] MIAO J M, XIAO F X, YANG H B, KHOO S Y, CHEN J, FAN Z. Hierarchical Ni-Mo-S nanosheets on carbon fiber cloth: A flexible electrode for efficient hydrogen generation in neutral electrolyte[J]. Science Advances, 2015, 1: 1500259.
[35] VOLPE L, BOUDART M. Compounds of molybdenum and tungsten with high specific surface area: ��. carbides[J]. Journal of Solid State Chemistry, 1985, 59: 348-356.
[36] MICHALSKY R, ZHANG Y J, PETERSON A A. Trends in the hydrogen evolution activity of metal carbide catalysts[J]. ACS Catalysis, 2014, 4: 1274-1278.
[37] WEIDMAN M C, ESPOSITO D V, HSU Y C, CHEN J G. Comparison of electrochemical stability of transition metal carbides (WC, W2C, Mo2C) over a wide pH range[J]. Journal of Power Sources, 2012, 202: 11-17.
[38] VRUBEL H, HU X. Molybdenum boride and carbide catalyze hydrogen evolution in both acidic and basic solutions[J]. Angewandte Chemie International Edition, 2012, 51: 12703-12706.
[39] �� ��, �ﺣ��, �� ��, ١����, лΰ��, �����. ���̼��������Ƭ���Ʊ���������[J]. �й���ɫ����ѧ��, 2015, 25(10): 2770-2776.
YANG Wei, SUN Hai-biao, YU Yang, TONG Ming-xing, XIE Wei-miao, LI Guo-hua. Preparation and electrocatalytic property of tungsten carbide nanoplate with mesoporosity[J]. The Chinese Journal of Nonferrous Metals, 2015, 25(10): 2770-2776.
[40] CHEN W F, WANG C H, SASAKI K, MARINKOVIC N, XU W, MUCKERMAN J T. Highly active and durable nanostructured molybdenum carbide electrocatalysts for hydrogen production[J]. Energy & Environmental Science, 2013, 6: 943-951.
[41] LIAO L, WANG S, XIAO J, GIRAULT H H. A nanoporous molybdenum carbide nanowire as an electrocatalyst for hydrogen evolution reaction[J]. Energy & Environmental Science, 2014, 7: 387-392.
[42] WAN C, LEONARD B M. Iron-doped molybdenum carbide catalyst with high activity and stability for the hydrogen evolution reaction[J]. Chemistry of Materials, 2015, 27: 4281-4288.
[43] XIONG K, LI L, ZHANG L, DING W, PENG L S, WANG Y, CHEN S G, TAN S Y, WEI Z D. Ni-doped Mo2C nanowires supported on Ni foam as a binder-free electrode for enhancing the hydrogen evolution performance[J]. Journal of Materials Chemistry A, 2015, 3: 1863-1867.
[44] BROCK S L, SENEVIRATHNE K. Recent developments in synthetic approaches to transition metal phosphide nanoparticles for magnetic and catalytic applications[J]. Journal of Solid State Chemistry, 2008, 181: 1552-1559.
[45] OYAMA S T. Novel catalysts for advanced hydroprocessing: Transition metal phosphides[J]. Journal of Catalysis, 2003, 216: 343-352.
[46] HENKES A F, VASQUEZ Y, SCHAAK R E. Converting metals into phosphides: A general strategy for the synthesis of metal phosphide nanocrystals[J]. Journal of the American Chemical Society, 2007, 129: 1896-1897.
[47] MUTHUSWAMY E, BROCK S L. Oxidation does not (always) kill reactivity of transition metals: Solution-phase conversion of nanoscale transition metal oxides to phosphides and sulfides[J]. Journal of the American Chemical Society, 2010, 132: 15849-15851.
[48] SUN J, LIU C, YANG P. Surfactant-free, large-scale, solution-liquid-solid (SLS) growth of gallium phosphide nanowires and their use for visible-light-driven hydrogen production from water reduction[J]. Journal of the American Chemical Society, 2011, 133: 19306-19309.
[49] FENG Z C, LIANG C H, WU W C, WU Z, van SANTEN R, LI C. CO adsorption on molybdenum phosphides: FT-IR spectroscopic and DFT studies[J]. Journal of Physical Chemistry B, 2003, 107: 13698-13702
[50] KANAMA D, OYAMA S T, OTANI S, COX D F. Photoemission and LEED characterization of Ni2P (0001)[J]. Surface Science, 2004, 552: 8-16.
[51] POPCZUN E J, MCKONE J R, READ C G, BIACCHI A J, WILTROUT A M, LEWIS N S, SCHAAK R E. Nanostructured nickel phosphide as an electrocatalyst for the hydrogen evolution reaction[J]. Journal of the American Chemical Society, 2013, 135(25): 9267-9270.
[52] POPCZUN E J, READ C G, ROSKE C W, ROSKE C W, SCHAAK R. Highly active electrocatalysis of the hydrogen evolution reaction by cobalt phosphide nanoparticles[J]. Angewandte Chemie International Edition, 2014, 53: 5427-5430.
[53] TIAN J Q, LIU Q, ASIRI A M, SUN X. Self-supported nanoporous cobalt phosphide nanowire arrays: An efficient 3D hydrogen-evolving cathode over the wide range of pH 0-14[J]. Journal of the American Chemical Society, 2014, 136: 7587-7590.
[54] JIANG P, LIU Q, LIANG Y, TIAN J, ASIRI A M, SUN X. A cost-effective 3D hydrogen evolution cathode with high catalytic activity: FeP nanowire array as the active phase[J]. Angewandte Chemie International Edition, 2014, 53(47): 12855-12859.
[55] LIANG Y, LIU Q, ASIRI A M, SUN X, LUO Y. Self-supported FeP nanorod arrays: A cost-effective 3D hydrogen evolution cathode with high catalytic activity[J]. ACS Catalysis, 2014, 4(11): 4065-4069.
[56] XIAO P, SK M A, THIA L, GE X, LIM R J, WANG J Y, LIM K H, WANG X. Molybdenum phosphide as an efficient electrocatalyst for the hydrogen evolution reaction[J]. Energy & Environmental Science, 2014, 7: 2624-2629.
[57] KIBSGAARD J, JARAMILLO T F. Molybdenum phosphosulfide: an active, acid-stable, earth-abundant catalyst for the hydrogen evolution reaction[J]. Angewandte Chemie International Edition, 2014, 53: 14433-14437.
XIONG Kun, GAO Yuan, ZHOU Gui-lin
(Engineering Research Center for Waste Oil Recovery Technology and Equipment, Ministry of Education, Chongqing Key Laboratory of Catalysis and Environmental New Materials, Chongqing Technology and Business University, Chongqing 400067, China)
Abstract: Hydrogen energy is a clean, high energy, environmentally friendly resource which is considered as one of the most promising candidates for replacing fossil fuels in the future. Electrochemically splitting water into hydrogen by renewable energy has attracted much attention due to its high efficiency, easy operation and no secondary pollution. Although the state-of-the-art catalyst for HER is a typical Pt-based catalyst, its scarcity and high cost prevent their widespread applications. Thus, it is highly desirable to exploit non-Pt HER catalysts with high activity, long-term stability and low cost. The recent advancements in the area of non-Pt HER catalysts with emphasis on introducing the exciting new research in the structure regulation of transition metal compounds and the understanding of the mechanisms of catalysts were reviewed. Meanwhile, the insights into the remaining challenges and research directions were proposed to shed light on future development of non-Pt HER catalysts.
Key words: water electrolysis; hydrogen energy; hydrogen evolution reaction; non-Pt catalyst
Foundation item: Project(21606028) supported by the National Natural Science Foundation of China; Project (KJ1500625) supported by the Science and Technology Project from Chongqing Education Commission, China; Project(2016-56-03) supported by the Scientific Research Foundation of Chongqing Technology and Business University, China
Received date: 2016-05-03; Accepted date: 2016-10-07
Corresponding author: XIONG Kun; Tel: +86-23-62768317; E-mail: kunxiong312@gmail.com
(�༭ �� ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(21606028)�������н�ί��ѧ�����о���Ŀ(KJ1500625)�����칤�̴�ѧ������������������Ŀ(2016-56-03)
�ո����ڣ�2016-05-03�������ڣ�2016-10-07
ͨ�����ߣ��� ������ʦ���绰��023-62768317��E-mail: kunxiong312@gmail.com

