DOI:10.19476/j.ysxb.1004.0609.2019.06.23
新型白钨矿分离羟肟酸捕收剂的分子设计
谭 鑫,张 华,王劲孚,方野平,佘小林,张建新
(中机国际工程设计研究院有限责任公司,长沙 410007)
摘 要:采用密度泛函理论模拟计算白钨矿(001)和萤石(111)解理面的结构特征及空间质点分布,并通过对羟肟酸与矿物表面吸附构型的分析,指出羟肟酸中非极性基对极性基官能团结构的影响会继而影响羟肟酸对白钨矿和萤石的选择性。论文基于该思路设计并合成一种新型的6-邻环己烷二甲酰亚胺基己基羟肟酸捕收剂(DIHHA)。通过核磁共振、元素分析以及红外光谱对合成产品进行表征,并对环己烷羟肟酸(CHA)进行了捕收剂的纯矿物浮选实验以及人工混合矿浮选实验。结果表明:DIHHA显示出比CHA更佳的选择性,是一种极具前景的白钨矿-萤石分离捕收剂。该实验结果与理论计算推测结果非常吻合,证明采用密度泛函理论模拟计算矿物晶体表面及捕收剂分子空间结构,并结合吸附构型分析,能够为捕收剂分子设计提供有效指导。
关键词:密度泛函理论;白钨矿;空间结构;羟肟酸
文章编号:1004-0609(2019)-06-1331-10 中图分类号:TD923 文献标志码:A
长期以来,白钨与萤石、方解石的含钙脉石矿物的浮选分离一直是困扰白钨矿资源高效利用的最关键问题。浮选主要利用矿物表面结构的疏水性差异进行分离,而白钨与萤石、方解石的表面活性质点都为钙离子,表面疏水性和化学活性相似,导致其相互间浮选分离困难。捕收剂是影响浮选分离最关键的因素之一,有关含钙矿物浮选分离捕收剂研究和应用的报道非常多。比如,脂肪酸类是就是其中应用最广泛的一类分离捕收剂[1-2]。胡岳华[3]对脂肪酸与白钨矿表面的化学反应机理进行了研究,通过动电位和吸附量测定、红外光谱分析、溶液化学计算和浮选实验证明,油酸钠能与矿物表面的金属离子发生化学吸附或表面反应,使矿物表面动电位发生负移,油酸钠在矿物表面的吸附行为条件与金属油酸盐生成的溶液化学条件具有相似性[4]。油酸捕收能力强,但是选择性较差,往往需要配合添加大量的选择性抑制剂才能使用。
羟肟酸则是目前含钙矿物浮选分离捕收剂研究和应用的热点[5-7]。WANG等[8]用烷基羟肟酸浮选超细白钨矿,发现烷基羟肟酸在碱性环境中具有较好的效果,并且随着烷基碳链的增长,适宜浮选的pH范围增大。羟肟酸的选择性强于脂肪酸的、毒性也比胂酸的小,是一种性能优良的钨锡捕收剂,但是其捕收能力相对较弱、用量也比较大、价格较高。虽然这些羟肟酸的研究取得了一定的成绩,但是整体上仍未能很好地解决目前含钙矿物分离难的问题。
近年来,钟宏等[9]报道了将一种新型的烃基内酰胺基羟肟酸引入含钙矿物浮选分离的研究。这种新型的烃基内酰胺基羟肟酸通过特定的连接基团联结,使得同样能够与金属离子形成螯合作用的酰胺基位于非极性基内部形成内酰胺基,由此获得了这种具有双特殊结构的烃基酰胺基羟肟酸。MOHAMADOU等[10]已经研究证明了通过该结合作用能够增强酰胺基氮氢原子之间的离子化作用,从而增强其余金属离子的结合能力。钟宏等[9]通过相关浮选试验的测试,表明酰胺基羟肟酸对白钨矿表现出极佳的选择性,证实了酰胺基羟肟酸是一种极具研发前途和应用前景的高性能羟肟酸捕收剂。然而,目前有关烃基酰胺羟肟酸的报道仍较少,酰胺羟肟酸还有很多方面值得继续深入全面的研究。
本文以白钨矿和萤石为例,采用密度泛函理论计算考察了白钨矿和萤石最常见解理面的表面结构特征,通过分析环己烷羟肟酸(CHA)与矿物表面的空间吸附模型,从空间匹配角度设计并合成一种新型的白钨矿分离捕收剂,即烃基二酰亚胺羟肟酸,并采用红外光谱、元素分析以及核磁共振验证了其结构。将这种新型的烃基二酰亚胺羟肟酸捕收剂应用于白钨与萤石的纯矿物和人工混合矿浮选试验。
1 实验
1.1 矿物样品与试剂
实验所用白钨矿纯矿物样品取自四川平武雪宝顶。萤石、方解石来自内蒙古锡林郭勒盟二连浩特市。所得实验样品经化学分析测试表明纯矿物品位分别为:白钨矿品位80.32%(WO3),纯度大于98%(质量分数);萤石纯度大于99%;方解石纯度大于96%。文中合成所用试剂均从专业试剂公司购买。浮选试剂均为分析纯配制,实验用水为蒸馏水。
1.2 矿物晶体及表面结构密度泛函理论计算方法
采用Material studio (MS)软件中的DMol3模块进行晶体、表面及浮选药剂的结构优化及量化性质计算。DMol3是一个MS软件中基于密度泛函理论的第一性原理计算模块。由于具有世界最高之一的量子力学计算速度,且能获较准确的计算结果,DMol3广泛应用于化学、医药、化工和固体物理及材料等各领域的计算和模拟。
本文量化计算的电子间的交换-关联效应通过基于广义梯度近似(GGA)的自旋极化泛函来考虑,采用由PERDEW和WANG[11]提出的泛函形式(PW91)作为交换相关函。PW91包含了关联函数的实空间截断以及较弱束缚的Beker交换函数,满足几乎所有已知的标度关系。矿物晶体优化,所选取的有效芯势为DFT 密度泛函数半核赝势(Sem-icorepseudopots, DSPP),代替重元素中非价电子层电子(1s22s22p6)。k点设置精度为高级,数值基组设置为DNP 3.5,轨道截断能精度为高级。自洽迭代收敛精度(SCF)为1×10-5 eV,能量收敛精度为1.0×10-5 eV,最大力收敛为2×10-3 eV/0.1 nm,最大位移收敛精度为5×10-4 nm,自洽迭代收敛精度(SCF)为高级。
1.3 产品合成实验及表征
6-邻苯二甲酰亚胺基己基羟肟酸:取5.6 g己内酰胺以及7.4 g邻苯二甲酸酐置于反应釜中,磁力搅拌,在180~200 ℃中反应18 h,减压蒸馏除去未反应原料,收集产品邻苯二甲酰胺己基酸。将上述产品置于圆底烧瓶中,加入过量甲醇(30 mL)以及溶剂甲苯(100 mL),缓慢滴加浓硫酸1.5 g,在110 ℃下回流8 h,反应所得混合物加入5%的Na2CO3溶液洗涤2次,除去反应的浓硫酸,洗涤2次,分液除水相,油相减压蒸馏除溶剂后得到酯产品。在酯产品中添加乙醇40 mL,盐酸羟胺3.5 g,添加烧碱4 g,在室温下搅拌8 h,得到粉色悬浊液,加入10%HCl(质量分数)溶液除过量的碱,干燥后最终得到淡红色固体。
采用元素分析以及核磁共振对获得的产品进行了表征。元素分析结果为,产品中C、H、O元素的占比(质量分数)分别为C 60.12%、H 5.75%、O 10.02%。对合成的BHHA进行核磁共振1H-NMR检测。检测结果如下:400 MHz 1H NMR (DMSO-d6): d 1.27-1.62(m, 6H), 2.35 (t, 2H), 3.68(t, 2H), 7.86(s, 4H), 7.12(s, H), 10.54(s, H)。
1.4 浮选实验方法
纯矿物及混合矿物浮选试验在XFG型挂槽式浮选机上进行,每次称取2.00 g矿样,加40 mL蒸馏水,调浆2 min后,用HCl、NaOH调节pH,搅拌2 min,然后依次加入调整剂、捕收剂和起泡剂,浮选5 min至泡沫中无产品。泡沫产品和槽内产品分别烘干、称量,计算回收率。浮选机转速为1680 r/min。
2 结果与讨论
2.1 矿物晶体结构特征分析
图1所示为白钨矿电子密度图和单晶胞图。白钨矿为四方晶系,为近八面体的四方双锥状,晶胞参数为,a0=b0=0.5243 nm,c0=1.1376 nm,α=β=γ=90°,Z=4,空间群为I4 1 /a。在晶胞中,W原子为中心原子,通过共价键与周围4个氧原子配位形成WO4四面体,W—O键长0.1778 nm。每个Ca2+与8个WO42-四面体中的八氧原子通过离子键形成八配位。Ca—O键长分别为0.2463 nm和0.2479 nm。WO4四面体是由Ca 联结起来,其一个二次对称轴与晶轴c平行,在c轴方向是一个被压扁的四面体,另外,2个二次对称轴与晶轴a、b相交45°,沿水平方向分布。白钨矿为离子晶体,其中的W原子与O原子以WO4四面体存在,W—O原子间不容易发生解离;而Ca2+相对容易从矿物晶体中解离,活泼性较高。
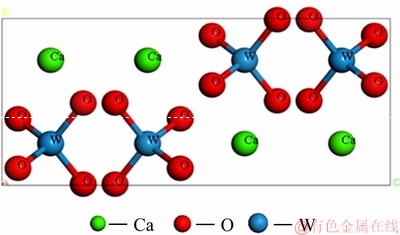
图1 白钨矿单晶胞模型图
Fig. 1 Unite cell model of scheelite
图2所示为白钨矿(001)面能带结构图和原子态密度图。白钨矿低能带区主要由O原子的3s和3p轨道组成,其中位于-19.0 eV附近的低能区极窄,这条能带为Ca2+上的3s轨道本征态,该轨道上的电子局域性非常强,因而能级较窄。-5.0~0 eV之间的价带区主要由O 2p轨道组成,该带区较平坦且非常密集,不具备较多的解读价值。导带区位于5.0 eV附近,以d轨道和p轨道成分为主,还包括较少的s轨道。晶体表面导带与价带之间的能隙为4.268 eV,属于绝缘体,晶体导电性较差。
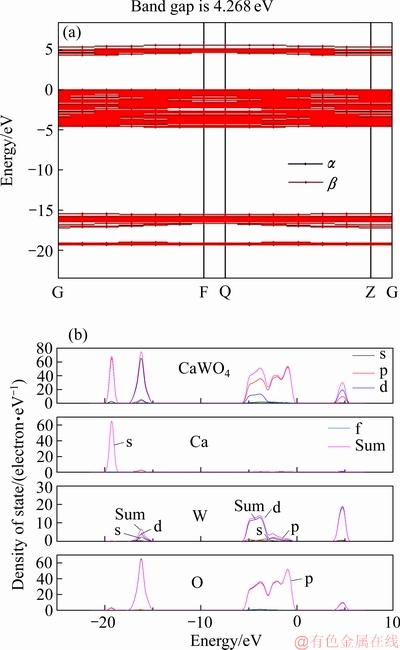
图2 白钨矿(001)表面能带结构图和原子态密度图
Fig. 2 Band structure(a) and density of state of atoms in scheelite (001) surface(b)
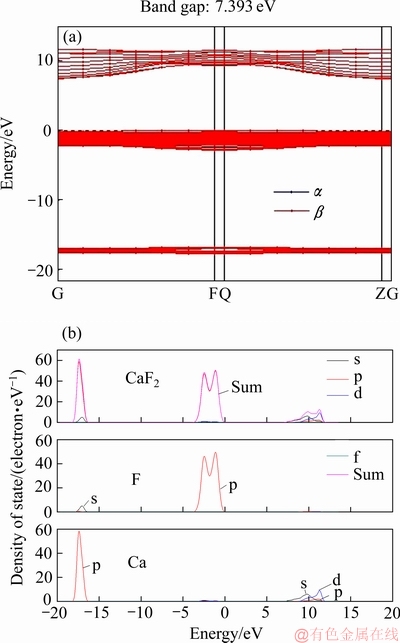
图3 萤石(111)面能带结构图和原子态密度图
Fig. 3 Band structure(a) and density of state of atoms in (111) cleavage surface of fluorite(b)
图3所示为萤石(111)面能带结构图和原子态密度图。图中,萤石晶体的能带低能区在-18.0~-16.0 eV 区域,基本为Ca 离子的3s轨道的本征态组成,而在费米能级附近的价带主要由F离子的2p轨道贡献,在7.5~12.5 eV附近的导带则主要由Ca2+中的s轨道和空d轨道组成,导带与价带间隙为7.393 eV,属于绝缘体。萤石晶体同样为离子晶体,Ca离子与F离子的态密度峰基本没有重叠,两者主要以离子键形式相互作用。
白钨矿及萤石表面的电子结构特征表明,Ca与其他元素的原子之间基本没有态密度峰的重叠,白钨矿和萤石是一种离子晶体,Ca与其他原子相互之间没有电子共用,主要失电子变成离子,通过离子键形成静电吸附。作为矿物表面活性位点的Ca2+极性较强,活泼程度相对较高,容易与阴离子捕收剂发生相互作用,因而,白钨矿可浮性较高。
由于Ca的还原性强难以与其余元素的原子发生电子共用,因此,在用阴离子浮选药剂对白钨矿进行浮选捕收作用时,捕收剂难以与白钨矿形成高选择性的共价结合,仅靠物理静电作用难以形成特定靶向进攻,因而导致捕收剂的选择性差,这是离子型矿物晶体面对的共同问题。
捕收剂与矿物间的作用其实是一种复杂的表面物理化学过程。两者的相互作用不仅仅受到表面化学反应的影响,更是受到了诸多譬如物理空间因素的影响。已经有众多研究表明,矿物表面活性质点密度及其空间方位分布是影响浮选药剂分子在矿物表面选择性吸附行为的关键的因素[12-16]。
因此,研究表面晶体化学结构,并针对两种矿物晶体表面Ca质点的密度和空间方位分布,设计出具有特殊空间匹配的捕收剂,实现矿物表面模型-捕收剂的空间锁钥匹配,有利于实现白钨矿与萤石的选择性浮选分离。
根据密度泛函理论计算结果,白钨矿(001)面和萤石(111)面表面正负电荷质点分布图见图4。
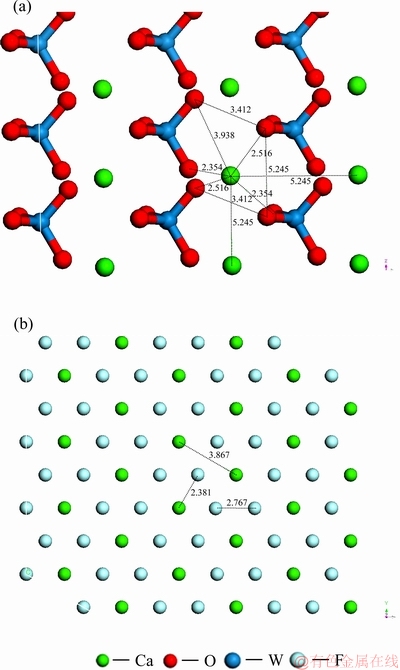
图4 白钨矿(001)面与萤石(111)面的正负电荷质点分布
Fig. 4 Distribution of positive and negative charge particles in scheelite (001) cleavage surface(a) and fluorite(111) cleavage surface(b) (Unit: 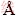 )
)
从图4可以看出,白钨矿(001)表面临近Ca质点之间的间距为0.5245 nm,WO4基团团簇模型的W质点距离为0.5245 nm,Ca质点与O质点的最临近距离为0.2345 nm,表面上最邻近的不同WO4基团之间O质点距离为0.3412 nm。而萤石(111)面Ca质点之间的间距为0.3867 nm,最邻近F质点的间距为0.2767 nm,Ca和F质点之间的距离为0.2381 nm。
2.2 矿物与羟肟酸的吸附构型分析
已经有大量的研究报道指出,羟肟酸在与矿物表面作用过程中,主要通过羟肟酸离子羟肟基中的两个带负电荷的O质点与矿物表面Ca2+形成闭合的螯合环状物结构。而具体的与矿物表面的结合形式,又可以因为矿物空间质点的差异以及捕收剂空间结构的差异而形成单核配位结构或双核配位结构[17-18]。
因此,本文以环己烷羟肟酸(CHA)为例,从空间角度分析羟肟酸与白钨和萤石吸附模型的结构差异,从而为捕收剂的设计提供指导。通过密度泛函理论计算表明环己烷羟肟酸的空间结构模型示意图如图5所示,环己烷羟肟酸分子和离子的空间结构参数分别见表1。
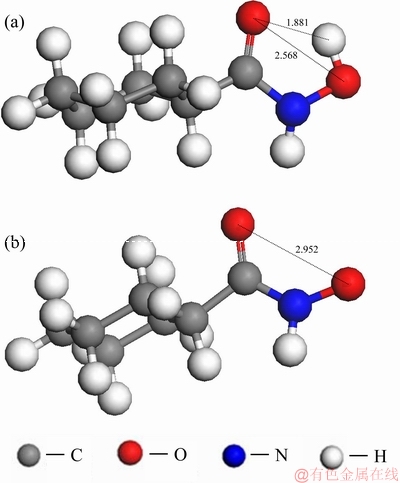
图5 CHA分子及阴离子模型示意图
Fig. 5 Schematic diagram of model of CHA molecule and CHA anion (Unit: )
因此,采用量子化学模型从空间角度,对羟肟酸与矿物晶体表面模型进行分析。假设当环己烷羟肟酸与白钨矿(001)表面Ca2+形成单核配位螯合物,且羟肟基中O质点与表面Ca2+之间的距离约为0.252 nm时(Ca和O原子的共价半径之和),吸附模型图见图6(图中略去非极性基团,后同),羟肟基中O质点与矿物表面负电荷的O质点的最临近距离为0.2683、0.2637、0.2632、0.2864 nm(以与O质点之间的距离最大位置摆放为原则)。以上数据说明,当环己烷羟肟酸离子在溶液中靠近白钨矿(001)表面形成单核双配位结构时,羟肟酸官能团中的O质点与矿物表面Ca质点的距离小于这2个O质点与表面负电荷较高的O质点的距离,即意味着该羟肟酸基团的空间结构与矿物表面空间结构呈单核配位匹配,有利于形成稳定的吸附结构。
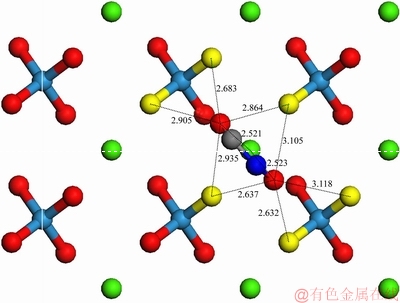
图6 环己烷-白钨矿(001)面单核双配位模型示意图
Fig. 6 Schematic diagram of bidentate model of CHA with scheelite (001) surface (Unit: )
假设环己烷羟肟酸与白钨矿(001)面形成双核双配位吸附,羟肟基中两个O质点分别与表面两个不同Ca2+的距离约为0.252 nm,吸附模型示意图见图7,此时羟肟酸O质点与表面临近O质点的距离为0.2443、0.2439、0.2423、0.2449 nm。以上结果表明,环己烷羟肟酸阴离子与白钨矿(001)面形成双配位结构时,羟肟基中带负电荷的O质点与矿物表面Ca质点的距离小于O质点与矿物表面负电荷的O质点距离,此时静电吸引力方向上的原子距离小于静电斥力方向上的原子距离,与单核双配位模型相比,即意味着羟肟酸阴离子的空间结构与矿物表面空间结构呈双核配位匹配相对较差,不利于形成稳定的吸附结构。
上述结果表明,羟肟酸在与白钨矿表面作用时,单核双配位的吸附构型相对更稳定,更容易形成单核配位结构产物。因此,单从白钨矿角度考虑,羟肟酸离子中2个O质点间的距离越短,2个O质点所在的直线则越远离矿物表面临近O质点所在的平面。此时,羟肟酸中O质点与矿物表面O质点的距离则越远。因此,进行羟肟酸分子设计时,当羟肟酸离子的2个O质点距离越短,则对白钨矿的捕收性能越强。
图7 CHA-白钨矿(001)面双核双配位模型示意图
Fig. 7 Schematic diagram of binuclear model of CHA with scheelite (001) surface (Unit: )
而对于萤石,假设当环己烷羟肟酸离子与萤石(111)表面Ca2+形成单核双配位螯合吸附,且羟肟基中带负电荷的O质点与表面Ca2+之间的距离约为0.252 nm时(吸附模型见图8)。羟肟基中O质点与矿物表面F-的最临近距离为0.2398、0.2161、0.2310、0.2248 nm(以O质点之间的距离最大位置摆放为原则)。此时,羟肟基中带负电荷的O质点与矿物表面Ca2+的距离小于O质点与矿物表面负电荷的F-距离,即静电吸引力方向上的原子距离小于静电斥力方向上的原子距离,意味着羟肟酸阴离子的空间结构与矿物表面空间结构呈双核配位匹配较差,不利于形成稳定的吸附结构。
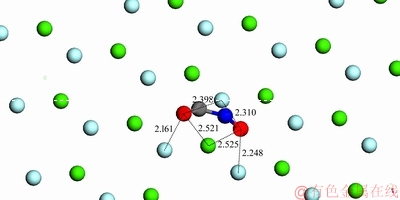
图8 CHA-萤石(111)面单核双配模型示意图
Fig. 8 Schematic diagram of bidentate model of CHA with fluorite (111) surface (Unit: )
假设当环己烷羟肟酸离子与萤石(111)表面Ca2+形成双核双配位吸附,羟肟基中2个带负电荷的O质点分别与表面2个不同Ca2+的距离约为0.252 nm(吸附模型见图9)。此时羟肟酸O质点与表面临近F-的距离为0.2654、0.2638、0.2618、0.2543 nm。羟肟酸官能团中的O质点与矿物表面Ca2+的距离小于这2个O质点与表面F-的距离,即表明该羟肟酸阴离子的空间结构与萤石(111)表面的空间结构呈单核双配位匹配,有利于形成稳定的吸附结构。
因此,与白钨矿不同,羟肟酸在与萤石矿物表面作用时,形成双核配位结构所受的斥力比单核配位结构所受斥力小,更容易形成稳定的双核配位结构产物。从萤石矿角度考虑,羟肟酸离子与矿物表面Ca2+呈双核配位螯合结构,若羟肟酸离子中2个O质点间的距离越短,由于这2个O质点距离总小于矿物表面Ca2+之间的距离,则2个O质点所在的直线则越靠近矿物表面临近O质点所在的平面。此时,羟肟酸中负电荷O质点与矿物表面F-的距离则越近。所以,当进行羟肟酸捕收剂分子设计时,当羟肟酸中2个O质点间距离越短,则越不容易形成稳定的结构,对萤石的捕收性能相对下降。
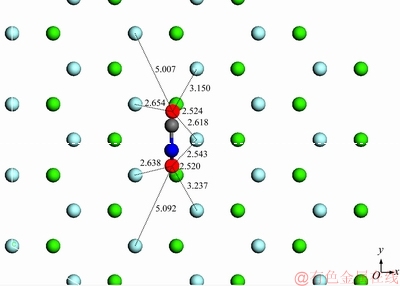
图9 CHA-萤石(111)面双核双配模型示意图
Fig. 9 Schematic diagram of binuclear model of CHA with scheelite (001) surface (Unit: )
2.3 新型捕收剂的设计及量化计算
上述分析可知,虽然白钨矿、萤石表面活性质点Ca2+一致,但其空间质点分布存在一定差异性,而羟肟酸与矿物表面的结合也呈现不同特点。因此,当羟肟酸官能团的空间结构分布能更匹配白钨矿的空间正负质点分布,犹如空间结构上锁与钥匙的匹配,则可以使捕收剂更好的结合。当羟肟酸捕收剂官能团中的2个O质点的空间距离能够缩短,从而有利于捕收剂结构能够与白钨矿矿物表面呈更好的空间匹配性,而与萤石矿表面匹配性相对较差。因而有利于对白钨与萤石的分离显示出更好的选择性。
基于前列结果,论文设计出一种特殊新型结构的6-邻环己烷二甲酰亚胺基己基羟肟酸(6-(1,3-dioxooctahydro-2H-isoindol-2-yl)-N-hydroxyhexanamide),简称环己烷二酰亚胺羟肟酸(DIHHA) 。首先进行量子化学结构优化,计算分子及其离子模型图见图10。结果表明,DIHHA分子电离后,其羰基O质点与羟基负电荷O质点的空间距离为0.2893 nm。2个负电荷O质点间空间距离比CHA离子更小,有利于与白钨矿表面的空间匹配,而与萤石的空间匹配差异性增大。
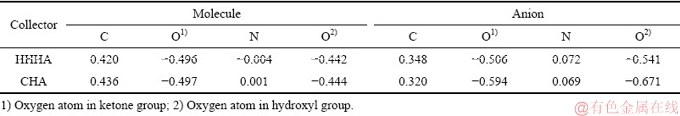
采用量子化学对DIHHA分子及离子进行了Mulliken电荷分析以及前线轨道分析。对比环己烷羟肟酸的结果如下表1所示。
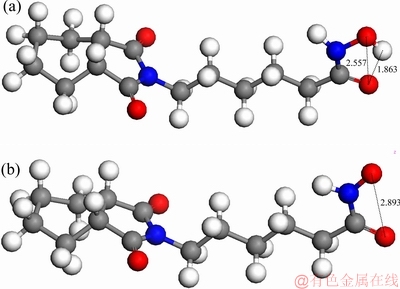
图10 DIHHA分子及阴离子模型示意图
Fig. 10 Schematic diagram of model of DIHHA molecule(a) and DIHHA anion ion(b) (Unit: )
从表1中可以看出,电离后羟基O以及羰基上O质点的负电荷量明显增加,因此,该2个O质点是与矿物表面Ca2+相作用的主要质点。CHA的官能团中,两个O质点负电荷增加明显,其中羟基O质点在失去氢原子后,Mulliken 电荷值增加-0.227,而羰基O质点的Mulliken电荷增加-0.097。而DIHHA分子电离后,羟基O质点上Mulliken 电荷值增加-0.099,羰基O质点增加-0.01。这可能主要是受到非极性基的影响,DIHHA中的六元环酰胺结构转移了部分负电荷,电离后二亚胺结构中原子的负电荷密度都有所增加。
表1 DIHHA和CHA分子及离子中官能团原子的所带电荷量
Table 1 Mulliken charge of selected atoms in CHA & DIHHA functional group
因此,从静电力学角度,环己烷羟肟酸中的氧原子具有更大的负电荷量,有利于与矿物表面发生更强的吸附作用,环己烷羟肟酸的捕收力应该较强。但是大量的浮选实践证明,捕收剂的捕收力越强,选择性则越差,且静电力作用没有选择性。因此,较强的静电反应力作用,易导致环己烷羟肟酸相对HHHA捕收剂的选择性差。
新型DIHHA分子的前线轨道特征值及能量值见表2。然而,前列已经分析了含钙矿物与捕收剂反应的前主要为静电控制反应,捕收剂的前线轨道对反应影响不大,因此不做过多分析。
表2 DIHHA分子前线轨道特征值及能量
Table 2 Frontier orbital eigenvalues and energies of DIHHA
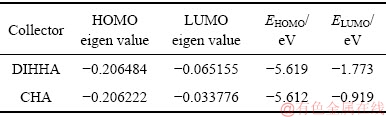
而从空间结构方面看,量子计算结果表明,电离后HHHA羟肟酸离子中,酮基氧中O18质点与羟基中O质点的空间距离为0.2893 nm,而环己烷中2个O质点的空间距离为0.2948 nm。
因此,根据前一节研究结果可知,在空间结构上,犹如锁与钥匙的匹配原理,DIHHA离子有利于与白钨矿(001)面呈更稳定的单核双配位吸附,DIHHA对白钨矿的捕收能力更强,而不利于与萤石(111)面呈稳定的双核双配位吸附,对萤石的捕收吸附能力弱于CHA。因而,DIHHA理论上具有比CHA更高的选择性。
2.4 合成及浮选验证实验
2.4.1 合成及表征
根据2.3节中方法合成了捕收剂DIHHA,并进行了元素分析、核磁共振以及红外光谱表征,产品的红外光谱图见图11。
从图11可以看出,环己烷二酰亚胺基羟肟酸(DIHHA)产品的红外光谱中3282及3234 cm-1处的吸收峰归属于羟肟基中的N—H键和O—H复合振动吸收峰。2956、2918、2823 cm-1吸收峰归属于非极性基中烃基—CH3、—CH2、—CH的伸缩振动。1712 cm-1处的尖峰归属于羰基C=O振动吸收峰,1390和1334 cm-1处的吸收峰归属于C—N键振动。
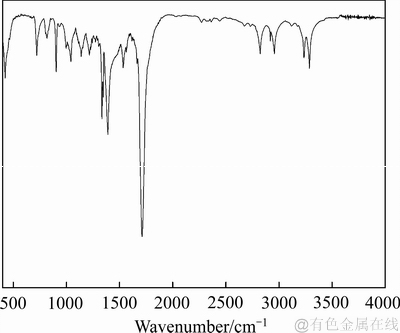
图11 DIHHA红外光谱
Fig. 11 IR spectrogram of DIHHA
2.4.2 纯矿物浮选对比试验
本实验中对比考察了新型二酰亚胺基羟肟酸DIHHA和传统环己烷基羟肟酸(CHA)作为捕收剂对白钨矿、萤石的浮选行为影响。其中捕收剂使用浓度为40 mg/L,起泡剂用量25 mg/L,辅助捕收剂F用量20 mg/L时,浮选pH=8.5对矿物回收率的影响如图12所示。
从图12可以看出,在浮选pH 4~9的范围内,HHHA对白钨矿的浮选捕收性能都强于对萤石的捕收性能。而环己烷则显示出对萤石更良好的捕收性能,且对萤石的适宜浮选pH范围为7~11,大于对白钨矿的适宜pH范围8~11。
在pH=8.5 的条件下,考察两种羟肟酸捕收剂浓度对两种矿石浮选回收率的影响。实验结果见图13。
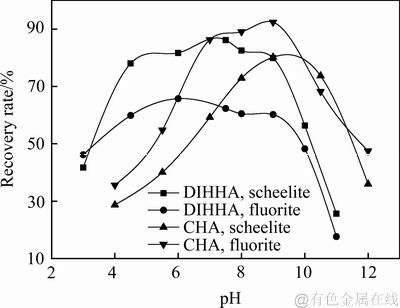
图12 pH对矿物浮选回收率的影响(CHA或DIHHHA浓度为40 mg/L)
Fig. 12 Effect of pH on recovery rate at 40 mg/L CHA or DIHHHA
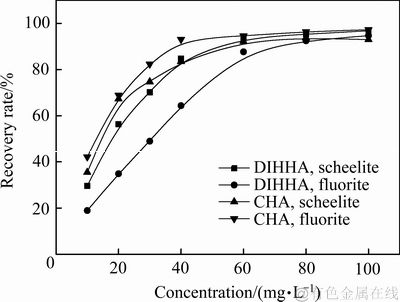
图13 捕收剂浓度对矿物浮选回收率的影响
Fig. 13 Influence of collector dose on recovery rate
从图13可以发现,在0~100 mg/L的浓度范围内,两种矿物的浮选回收率都随着两种捕收剂浓度的增加,但是当浓度超过80 mg/L时,浮选回收率的增加已经不明显。在10~60 mg/L范围内,CHA对萤石和白钨矿的捕收能力强于DIHHA的,而DIHHA对白钨矿的捕收能力明显强于对萤石的。整体上,DIHHA对白钨矿显示出更好的选择性,CHA对含钙矿物的捕收能力强于DIHHA。该浮选实验结果与先前的理论计算推断结果相一致。
2.4.3 人工混合矿浮选对比试验
采用白钨与萤石质量比2:3的人工混合矿进行了对比实验,每次混合矿取样为2.5 g,起泡剂用量为25 mg/L,调整剂水玻璃用量100 mg/L,捕收剂用量均为60 mg/L,辅助捕收剂F用量25 mg/L,在pH值=8.5的条件下进行,最终实验结果如表3所示。
表3 人工混合矿捕收剂浮选对比实验结果
Table 3 Contrast of flotation results of artificial mixed ores
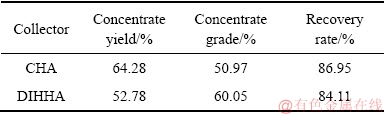
由表3可知,CHA具有较强的捕收能力,其产品的产率更高,但是DIHHA显示出对白钨矿和萤石浮选分离的更好选择性,在两者精矿产品回收率相差不大的情况下,使用DIHHA作为捕收剂的精矿产品的品位比使用CHA的精矿品位高出近10%。证明了论文前列理论研究结果的正确性。
3 结论
1) 白钨矿(001)面和萤石(111)面晶体表面结构特征表明,这两种晶体属于离子晶体,Ca2+为表面活性质点,主要以静电作用与捕收剂阴离子相作用。
2) 受矿物表面结构及羟肟酸阴离子空间结构的影响。羟肟酸与白钨矿(001)面主要呈单核双配位结构吸附,而与萤石(111)面呈双核双配位吸附。当羟肟酸离子中,两个带负电荷的O质点的空间距离越短,与白钨矿(001)面质点的单核配位空间匹配性更佳,而与萤石(111)面质点的双核双配位空间匹配性越差,因而对白钨矿的选择性会更好。
3) 设计和合成的新型环己烷基二酰亚胺羟肟酸从空间结构上分析具有比环己烷羟肟酸更好的选择性。最终的浮选实验验证了上述结果。采用量子化学模拟计算,结合捕收剂及矿物表面的空间结构特征分析,从空间锁钥匹配角度能够为新型白钨矿羟肟酸捕收剂的分子设计提供指导。
致谢:
本论文的完成感谢北京矿冶研究总院矿物工程研究所和东北大学资源与土木工程学院在MS 7.0软件所有密度泛函理论计算上提供的支持。
REFERENCES
[1] HANUMANTHA R K, FORSSBERG K S E. Mechanism of oleate interaction on salt-type minerals Part Ⅲ. Adsorption, zeta potential and diffuse reflectance FT-IR studies of scheelite in the presence of sodium oleate[J]. Colloids and Surfaces, 1991, 54: 161-187.
[2] ANTTI B M, FORSSBERG E. Pulp chemistry in calcite flotation. modelling of oleate adsorption using theoretical equilibrium calculations[J]. Minerals Engineering, 1989, 2(1): 93-109.
[3] 胡岳华. 溶液化学计算及图解法与盐类矿物浮选行为研究[D]. 长沙: 中南大学, 1989.
HU Yue-hua. Solution chemistry calculation and graphic method and flotation behavior of salt minerals[D]. Changsha: Central South University, 1989.
[4] HANUMANTHA R K, FORSSBERG K S E. Mechanism of fatty acid adsorption in salt-type mineral flotation[J]. Minerals Engineering, 1991, 4(7/11): 879-890.
[5] SREENIVAS T, PADMANABHAN N P H. Surface chemistry and flotation of cassiterite with alkyl hydroxamates[J]. Colloids and Surfaces A: Physicochemical and Engineering Aspects, 2002, 205: 47-59.
[6] 韩兆元, 管则皋, 卢毅屏. 组合捕收剂回收某钨矿的试验研究[J]. 矿冶工程, 2009(1): 50-52.
HAN Zhao-yuan, GUAN Ze-Hao, LU Yi-ping. Experimental study on recovery of a tungsten ore by combined collector[J]. Mining and Metallurgy Engineering, 2009(1): 50-52.
[7] 周晓彤, 杨应林, 汤玉和. 某难选黑白钨共生矿试验研究[J]. 中国钨业, 2012, 27(1): 27-40.
ZHOU Xiao-Tong, YANG Ying-lin, TANG Yu-he. Test research of one black and white tungsten ore refractory [J]. China tungsten, 2012, 27(1): 27-40.
[8] WANG Pei-pei, QIN Wen-qing, REN Liu-yi, WEI Qian, LIU Rui-zeng, YANG Cong-ren, ZHONG Shui-ping. Solution chemistry and utilization of alkyl hydroxamic acid in flotation of fine cassiterite[J]. Transactions Nonferrous Metals Society of China, 2013, 23(6): 1789-1796.
[9] 钟 宏, 王 帅, 邓兰青, 黄志强, 刘广义, 曹占芳. 6-芳基酰胺基己基羟肟酸捕收剂及其制备和应用方法. CN 103301953 A[P]. 2013-09-18.
ZHONG Hong, WANG Shuai, DENG Lang-qing, HUANG Zhi-qiang, LIU Guang-yi, CAO Zhan-fang. The reparation and application method of 6- aryl amide hexyl hydroxamic acid collector. CN 103301953 A[P]. 2013-09-18.
[10] MOHAMADOUA, MOREAU J, DUPONT L, WENGER E. Equilibrium and structural studies of copper(Ⅱ) complexes with tridentate ligand containing amide, pyrazyl and pyridyl nitrogen donors: effect of anions coligands on crystal structures of copper(Ⅱ) complexes[J]. Inorg Chim Acta, 2012, 383: 267-276.
[11] PERDEW J P, WANG Y. Accurate and simple analytic repesentation of the electron-gas correlation energy[J]. Physical Review A, 1992, 45(13): 13244-13249.
[12] MOON K S, FUERSTENAU D W. Surface crystal chemistry in selective flotation of spodumene from other aluminosilicates[J]. International Journal of Mineral Processing, 2003, 72(1): 11-24.
[13] RAI B, SATHISH P, TANWAR J, PRADIP, MOON K S, FUERSTENAU D W. A molecular dynamics study of the interaction of oleate and dodecylammonium chloride surfactants with complex aluminosilicate minerals[J]. Journal of Colloid and Interface Science, 2011, 362(2): 510-516.
[14] PRADIP, RAI B, RAO T K, KRISHNAMURTHY S, VETRIVEL R, MIELCZARSKI J, CASES J M. Molecular modeling of interactions of diphosphonic acid based surfactants with calcium minerals[J]. Langmuir, 2002, 18(3): 932-940.
[15] MIELCZARSKI E, MIELCZARSKI J A, CASES J M. Molecular recognition effect in monolayer formation of oleate on fluorite[J]. Langmuir, 1998, 14(7): 1739-1747
[16] HIRVA P, TIKKA H K. Ab initio study on the interaction of anionic collectors with calcite and dolomite surfaces[J]. Langmuir, 2002, 18(12): 5002-5006.
[17] ZHAO Gang, ZHONG Hong, QIU Xian-yang, WANG Shuai, GAO Yu-de, DAI Zi-lin, HUANG Jian-ping, LIU Guang-yi . The DFT study of cyclohexyl hydroxamic acid as a collector in scheelite flotation[J]. Minerals Engineering, 2013, 49(8): 54-60.
[18] TAN Xin, HE Fa-yu, SHANG Yan-bo, YIN Wan-zhong. Flotation behavior and adsorption mechanism of (1-hydroxy-2-methyl-2-octenyl) phosphonic acid to cassiterite[J]. Transactions of Nonferrous Metals Society of China, 2016, 26(9): 2469-2478.
Molecular design of a new hydroxamic acid collector for scheelite flotation separation
TAN Xin, ZHANG Hua, WANG Jin-fu, FANG Ye-ping, SHE Xiao-lin, ZHANG Jian-xin
(China Machinery International Engineering Design and Research Institute Co., Ltd., Changsha 410007, China)
Abstract: According to the structure characteristics and spatial distribution of scheelite (001) and fluorite (111) cleavage surfaces calculated by the density functional theory, together with the analysis of adsorption model of hydroxamic acid with crystal surfaces, the selectivity of hydroxamic acid collector for scheelite flotation separation was thought to be affected by the structure of the polar group, which was affected by the non-polar groups. Based on this idea, a new type of (6-(1,3-dioxooctahydro-2H-isoindol-2-yl)-N-hydroxyhexanamide) (DIHHA) collector was designed and synthesized. The synthetic products were characterized by NMR, elemental analysis and infrared spectroscopy. The scheelite and fluorite pure mineral flotation and artificial mixed minerals flotation experiments of DIHHA were studied by compared with cyclohexane hydroxamic acid (CHA). The results show that DIHHA has better selectivity than CHA and is a promising new collector for flotation separation of scheelite and fluorite. The experimental results are in good agreement with the theoretical calculations, which indicates that the simulation calculation of mineral crystal surface and collector molecular structure by density functional theory, combined with the adsorption configuration analysis, can provide effective guidance for the design of collector molecules.
Key words: density functional theory; scheelite; spatial structure; hydroxamic acid
Received date: 2018-01-26; Accepted date: 2019-05-09
Corresponding author: TAN Xin; Tel: +86-13581507345; E-mail: tanxin0222@hotmail.com
(编辑 龙怀中)
收稿日期:2018-01-26;修订日期:2019-05-09
通信作者:谭 鑫,工程师,博士;电话:13581507345;E-mail:tanxin0222@hotmail.com

