网络首发时间: 2015-04-07 09:26
稀有金属 2016,40(01),92-96 DOI:10.13373/j.cnki.cjrm.2016.01.015
Ni对bcc-Fe/ε-Cu界面影响的第一性原理研究
王海燕 高雪云 任慧平 李德超 刘宗昌
内蒙古科技大学材料与冶金学院
内蒙古白云鄂博矿多金属资源综合利用重点实验室
中冶东方工程技术有限公司长材事业部
摘 要:
采用基于密度泛函理论(DFT)的第一性原理,研究了Ni对bcc-Fe/ε-Cu界面结合的影响。建立了ε-Cu在bcc-Fe的析出模型,选取界面两侧不同阵点位置,计算Ni在不同位置的偏聚能,分析了Ni在界面区域的占位倾向,在此基础上探究了Ni对bcc-Fe/ε-Cu界面结合的影响。利用Rice-Wang热力学模型的计算表明,当Ni原子处于偏聚能最低的位置时,能够强化界面的结合。而界面分离功计算结果显示,Ni偏聚于bcc-Fe/ε-Cu界面后,界面分离功由279.8 m J・m-2增加到286.7 m J・m-2,表明Ni偏聚后会使界面体系更加稳定。Ni偏聚于界面后对界面区域的电子结构也产生一定影响,差分电荷密度显示,与纯bcc-Fe/ε-Cu界面相比,Ni偏聚后会在其周围聚集较多的电子,且Ni与相邻原子之间电子云方向性更为明显;同时,Ni也使近邻Cu和Fe原子的态密度(DOS)向成键态偏移,这使得Ni偏聚加强了bcc-Fe/ε-Cu界面的结合,使界面区更为稳定。
关键词:
Ni;bcc-Fe;Cu;第一性原理;
中图分类号: TG142.1
作者简介:王海燕(1975-),女,内蒙古包头人,博士研究生,副教授,研究方向:优势资源金属材料研究与开发;电话:13284712228;E-mail:windflower126@163.com;
收稿日期:2014-08-22
基金:国家自然科学基金项目(50361001)资助;
First-Principles Study of bcc-Fe/ε-Cu Interface with Different Ni Contents
Wang Haiyan Gao Xueyun Ren Huiping Li Dechao Liu Zongchang
School of Material and Metallurgy,Inner Mongolia University of Science and Technology
Key Laboratory of Integrated Exploitation of Bayan Obo Multi-Metal Resources
Beris Engineering and Research Corporation
Abstract:
The influence of Ni on the bcc-Fe/ε-Cu interface was investigated by means of first-principles calculations based on the density functional theory( DFT). To identify the most preferred energetic site for Ni atom,the atom model of ε-Cu precipitation in bccFe was built,the segregation energies of Ni atom at different sites of bcc-Fe/ε-Cu interface was calculated,and the effect of Ni on the interfacial binding was discussed as well. From the calculations based on Rice-Wang model,it could be found that Ni atom induced enhancement effect on the interface when in the lowest segregation energy state. And the work of adhesion of the interface with Ni segregation was 286. 7 m J・m-2,higher than the Ni free one that was 279. 8 m J・m-2,which indicated that Ni segregation made the interface system more stable. The result of the calculated electronic structure showed that when Ni segregated at the bcc-Fe/ε-Cu interface,the electrons around Ni increased,and the orientation of electron clouds between Ni and its neighboring atoms was enhanced; at the meantime,the density of atoms( DOS) of Cu and Fe atom near the Ni atom moved to the bond state somewhat,which made the interface more stable.
Keyword:
Ni; bcc-Fe; Cu; first-principles;
Received: 2014-08-22
随着超低碳、超纯净钢冶炼和微合金化等技术的发展,以美国开发的含铜HSLA系列船体钢为标志,铜在新一代钢铁材料中的研究与应用愈来愈受到材料科学研究者的的广泛关注[1,2,3]。含铜高强低合金钢具有高强度、高韧性、良好的可焊性、优良的耐蚀性等多种综合性能。Cu加入钢中后,依靠Cu的时效析出作用,在bcc-Fe基体的析出可使钢得到显著强化,同时保持较高的塑韧性。因此,认识Cu在钢中的脱溶析出过程成为分析Cu时效强化的重要依据。
目前,国内外学者对含铜钢中富铜相的时效析出行为进行了大量研究[4,5,6,7,8]。结果表明,含铜纳米相结构对钢的力学性能具有直接影响。通过合适的热机械处理,可在含铜HSLA钢中获得具有特定结构的纳米相,其结构及热稳定性与钢的化学成分和热处理工艺密切相关。其中,Ni,Al等溶质可溶于富Cu纳米团簇中或析出到界面,在大幅度提高钢的强度的同时,使其保持良好的塑韧性[9]。对Fe-Cu-Ni合金的研究发现,在Cu析出过程中,Ni偏聚于bcc-Fe / ε-Cu析出物的界面区域,并且有利于加速Cu的析出[10]。然而,由于Cu的沉淀贯序较为复杂,且铜在钢中的析出物极为细小,沉淀粒子对钢强度的贡献较难模拟。目前Ni对Cu析出相的析出贯序及影响机制尚鲜有系统的研究报道。
本文将利用基于密度泛函理论的第一性原理,通过计算Ni在bcc-Fe/ε-Cu界面的偏聚能,确定Ni在界面区域的占位倾向,在此基础上讨论Ni对bcc-Fe/ε-Cu界面结合的影响,从电子结构层次探究研究Ni对Cu原子析出强化行为的影响,为含铜新型HSLA钢的成分合金化设计提供理论依据。
1计算方法与结构模型
本文采用基于密度泛函理论(DFT)框架下的Vienna Ab-initio Simulation Package(VASP)软件包[11]。选择投影缀加波(PAW)方法的广义梯度近似(GGA),截断能量为350 e V。布里渊区积分采用Monhkorst-Pack特殊k网格点方法,选取了6×6×1的k网格。计算中的能量收敛标准为能量小于10-4e V,每个原子的剩余力小于0.001 e V・nm-1。bcc-Fe/ε-Cu界面结构模型如图1所示,bcc-Fe和fcc结构的ε-Cu之间为K-S位向关系:(111)ε-Cu//(110)bcc-Fe,ε-Cu为fcc结构,该析出物与bcc-Fe基体之间存在K-S位向关系, [12]。图1中上半部分较小圆球表示Cu原子,下半部分较大圆球表示Fe原子,颜色深浅表示不同的层,模型共包含56个Cu原子和64个Fe原子。本文用Ni原子替换图1中位置0的Fe原子,以模拟Ni固溶于bcc-Fe基体内部。位置1,2,3和4,5,6分别为界面bcc-Fe侧和Cu析出物侧的不同点阵位置。
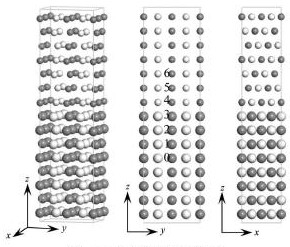
图1 bcc-Fe/ε-Cu界面结构模型Fig. 1 Atomic configuration of bcc-Fe / ε-Cu interface
2 计算结果与讨论
2.1 Ni在界面的偏聚能计算
在研究Ni对bcc-Fe/ε-Cu界面的影响之前,本文通过计算Ni在界面模型不同点阵位置的偏聚能来确定Ni的优先占位。偏聚能通过以下公式计算得到[13,14]:
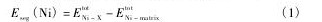
式中,EtotNi-X为Ni占据图1界面模型中不同点阵位置时体系的总能量,EtotNi-matrix为Ni占据bcc-Fe基体内(位置0)时体系的总能量。若Ni在某一点阵位置的偏聚能为负值,表示Ni趋向于从基体向该位置扩散;若偏聚能为正值,则表示Ni趋向于在bcc-Fe基体内部固溶。Ni在bcc-Fe/ε-Cu界面不同位置的偏聚能计算结果如图2所示。
由图2 可以看出,随着偏聚位置从bcc-Fe基体向界面不断推移,Ni的偏聚能不断减小,到位置4 时达到最小值- 0. 57 e V; 随着偏聚位置继续向Cu析出物深入,偏聚能不断增大,到位置6 时达到0. 02 e V。偏聚能的计算结果表明,Ni易于偏聚在位置4,下文将针对Ni偏聚于位置4 后对界面的影响展开研究。
2.2 Ni对界面结合的影响
Rice-Wang热力学模型广泛用于描述杂质对界面结合的影响[15,16]。基于该模型,可通过计算杂质分别偏聚在界面和表面时的结合能之差,来判断杂质对界面的钝化或韧化机制:
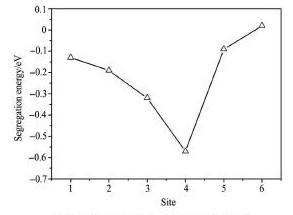
图2 Ni处于界面不同位置时的偏聚能Fig. 2 Segregation energies of Ni atom at different interface sites

其中,ΔEIF和 ΔEFS分别为杂质偏聚在界面和自由表面时的结合能,通过下式得到:
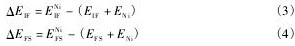
式中,EIFNi和EFSNi分别为界面和自由表面在掺杂Ni后的总能量,EIF和EFS分别为晶界和表面在不含杂质时的总能量,ENi为Ni原子的总能量。
将( 3) 与( 4) 式代入( 2) 式后,得到式( 5) :
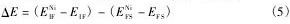
若 ΔE为正值,表示掺杂后界面断裂功减少,杂质元素削弱界面的结合; ΔE为负值,则表示杂质的偏聚强化界面的结合。通过式( 5) 的计算,得到Ni偏聚于图1 所示位置4 时的 ΔE为- 0. 32e V,说明Ni偏聚能够强化bcc-Fe / ε-Cu界面的结合。
界面分离功Wad表示在忽略弹性性能、缺陷尺寸和扩散等条件下,将界面分离成两个自由表面所需做的功[17,18],利用下式计算:
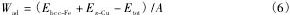
其中,Ebcc-Fe为模型中的Cu用1. 0 nm真空层取代并完全结构优化后的总能量,Eε-Cu为将模型中bccFe用1. 0 nm真空层取代并完全结构优化后的总能量,Etot表示界面体系的总能量,A表示界面面积。
经计算,界面在Ni偏聚前后的分离功分别为279. 8 和286. 7 m J・m- 2,前者与Medvedeva等[19]报道的276 m J・m- 2较为接近,证明本模型的计算结果是合理的。由于Ni偏聚后,使bcc-Fe /ε-Cu的界面分离功由279. 8 m J・m- 2增加到286. 7 m J・m- 2,表明Ni的加入可使界面更加稳定[20]。
2.3 Ni对界面电子结构的影响
为了进一步分析Ni对bcc-Fe/ε-Cu界面结合的影响,计算了Ni在图1所示位置4偏聚前后的差分电荷密度。为了清晰显示Ni影响区域的电子结构变化,截取图1所示界面模型中包含位置4的(100)和(010)面的差分电荷密度分布,如图3所示,图中深色表示电荷缺失,浅色表示电荷富集。图3(a)和(b)中,Cu1原子为图1中所示位置4的Cu原子,在图3(c)和(d)中被Ni原子替代。
由图3( a) 与( b) 可以看出,不含有Ni原子的界面中,Fe和Cu原子周围的电子有所减少,而公共区域则有电子富集。图3( c) 与( d) 显示,当Ni偏聚于界面后,Ni周围的电子显著富集,且Ni与周围的Fe和Cu原子之间的电子云方向性更为明显,电子云方向性的增强表明其成键能力有所增强。以上这些变化均表明,Ni偏聚后加强了bccFe / ε-Cu界面的结合[21]。
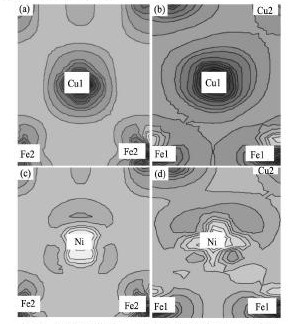
图3 Ni偏聚前后bcc-Fe/Cu界面的差分电荷密度Fig. 3Valence charge density differences of bcc-Fe / ε-Cu interface systems before and after Ni segregation
(a)(100)plane for bcc-Fe/ε-Cu;(b)(010)plane for bccFe/ε-Cu;(c)(100)plane for bcc-Fe/ε-Cu-Ni segregation;(d)(010)plane for bcc-Fe/ε-Cu-Ni segregation
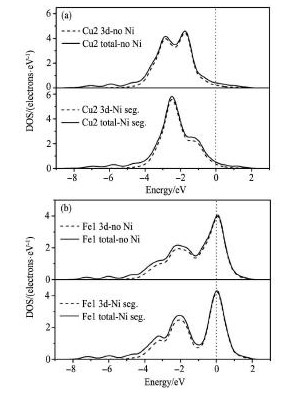
图4 bcc-Fe/ε-Cu界面偏聚Ni前后偏聚区Cu原子和Fe原子的态密度Fig. 4 Densities of state of Cu( a) and Fe( b) of bcc-Fe / ε-Cu interface with Ni free and doped
界面在偏聚Ni前后,偏聚位置周围Cu和Fe原子的态密度如图4 所示,图4 中的合金原子与图3 中的对应,Fe1 原子和Fe2 原子的态密度相同,本文仅给出Fe1 的态密度图。
图4 表明,Fe和Cu的态密度主要由3d轨道电子贡献。Ni偏聚到界面对Cu2 的态密度有明显的影响,使- 1 ~ - 2 e V能量范围的峰值降低的同时,使- 2 ~- 4 e V范围的态密度显著升高。从图4( b) 也可以看出,Ni使Fe1 原子在- 1 ~ - 3 e V能量范围的态密度有所升高。以上分析表明,Ni偏聚到界面后,使周围Cu和Fe原子态密度峰值向成键态偏移,使界面的结合更为稳定。
3 结论
利用密度泛函理论研究了Ni在bcc-Fe /ε-Cu析出物界面偏聚对界面结合的影响。Ni在铁基体和析出物中不同位置的偏聚能的计算结果表明,Ni易于偏聚在界面处与bcc-Fe相邻的 ε-Cu侧。根据Rice-Wang热力学模型,Ni在界面偏聚后,可以强化界面的结合。Ni偏聚后界面的分离功从279. 8增加到286. 7 m J・m- 2,这验证了Ni在界面的偏聚可以增强界面的稳定性。电子结构的计算结果表明,Ni偏聚于界面后,其周围聚集了较多的电子,且与相邻原子之间电子云方向性更为明显; 同时,Ni也使相邻的Cu和Fe原子的态密度向成键态偏移,从而使界面区更为稳定。
参考文献
[1] Othen P J,Jenkins M L,Smith G D W.High-resolution electron microscopy studies of the structure of Cu precipitations inα-Fe[J].Philosophical Magazine A,1994,70(1):1.
[2] Deschamps A,Militzer M,Poole W J.Precipitation kinetics and strengthening of a Fe-0.8 wt%Cu alloy[J].ISIJ International,2001,41(2):196.
[3] Goodman S R,Brenner S S,Low R J J.An FIM-atom probe study of the precipitat ion of copper from iron 1.4at pct copper[J].Metallurgical Transactions,1973,4(10):2363.
[4] Fine M E,Isheim D.Origin of copper precipitation strengthening in steel[J].Scripta Materialia,2005,53(1):115.
[5] Nogiwa K,Nita N,Matsui H.Quantitative analysis of the dependence of hardening on copper precipitate diameter and density in Fe-Cu alloys[J].Journal of Nuclear Materials,2007,367-370:392.
[6] Miller M K,Russell K F,Pareige P,Starink M J,Thomson R C.Low temperature copper solubilities in Fe-Cu-Ni[J].Materials Science and Engineering A,1998,250(1):49.
[7] Zhang Z W,Liu C T,Wang X L,Miller M K,Ma D,Chen G,Williams J R,Chin B A.Effects of proton irradiation on nanocluster precipitation in ferritic steel containing fcc alloying additions[J].Acta Materialia,2012,60(6-7):3034.
[8] Heo Y U,Kim Y K,Kim J S,Kim J K.Phase transformation of Cu precipitates from bcc to fcc in Fe-3Si-2Cu alloy[J].Acta Materialia,2013,61(2):519.
[9] Zhang C,Enomoto M.Study of the influence of alloying elements on Cu precipitation in steel by non-classical nucleation theory[J].Acta Materialia,2006,54(16):4183.
[10] Isheim D,Gagliano M S,Fine M E,Seidman D N.Interfacial segregation at Cu-rich precipitates in a highstrength low-carbon steel studied on a sub-nanometer scale[J].Acta Materialia,2006,54(3):841.
[11] Chen S,Lu J S,Xie M,Pan Y,Hu J Q,Wang S.First-principle investigation on problem of ruthenium allotropic phase stability[J].Chinese Journal of Rare Metlals,2015,39(12):1095.(陈松,陆建生,谢明,潘勇,胡洁琼,王松.金属钌同素异构体问题的第一性原理计算研究[J].稀有金属,2015,39(12):1095.)
[12] Ryoichi Monzen,Kenichi Takada,Chihiro Watanabe.Coarsening of spherical Cu particles in an bcc-Fe matrix[J].ISIJ International,2004,44(2):442.
[13] Zhang S J,Kontsevoi O Y,Freeman Arthur J,Olson G B.First principles investigation of zinc-induced embrittlement in an aluminum grain boundary[J].Acta Materialia,2011,59(15):6155.
[14] Christensen M,Angeliu T M,Ballard J D,Vollmer J,Najafabadi R,Wimmer E.Effect of impurity and alloying elements on Zr grain boundary strength from firstprinciples computations[J].Journal of Nuclear Materials,2010,404(2):121.
[15] Liu W G,Han H,Ren C L,He X J,Jia Y Y,Wang S,Zhang W,Li Z J,Zhou X T,Zou Y,Huai P,Xu H J.First-principles study of intergranular embrittlement induced by Te in the Ni R 5 grain boundary[J].Computational Materials Science,2014,88(1):22.
[16] Wu Y X,Wang Y M.First-principles study on the ductility effect of zirconium and its distinct behavior from boron to restrain hydrogen-induced embrittlement in NiNi3Al alloys[J].Journal of Materials Scienc and Technology,2008,24(2):165.
[17] Donald J S,Louis G H J,James B A.First-principles study of metal-carbide/nitride adhesion:Al/VC vs.Al/VN[J].Acta Materialia,2002,50(3):619.
[18] Wang H Y,Zhang S,Li D J,Liu S Y.The simulation of adhesion,stability,electronic structure of W/Zr B2interface using first-principles[J].Surface and Coatings Technology,2013,228(S1):583.
[19] Medvedeva N I,Murthy A S,Richards V L,Van Aken D C,Medvedeva J E.First principle study of cobalt impurity in bcc Fe with Cu precipitates[J].Journal of Materials Science,2013,48(3):1377.
[20] Donald J S,Louis G H J,James B A.Adhesion,stability,and bonding at metal/metal-carbide interfaces:Al/WC[J].Surface Science,2002,498(3):321.
[21] Wang H Y,Gao X Y,Ren H P,Zhang H W,Tan H J.First-principles characterization of lanthanum occupying tendency inα-Fe and effect on grain boundaries[J].Acta Physica Sinica,2014,63(14):148101.(王海燕,高雪云,任慧平,张红伟,谭会杰.稀土La在α-Fe中占位倾向及对晶界影响的第一性原理研究[J].物理学报,2014,63(14):148101.)

