���±�ţ�1004-0609(2016)-11-2403-09
�ռ�ṹ����������ܴ��ṹ�͵������ʵ�Ӱ��
�� ��1, 2���½���1, 2��������1, 2���Դ仪3
(1. ������ѧ ��Դ��ұ��ѧԺ������ 530004��
2. ������ѧ ������У���﹤���ص�ʵ���ң����� 530004��
3. ������ѧ ���Ͽ�ѧ�빤��ѧԺ������ 530004)
ժ Ҫ�������ܶȷ������ۣ����㷽Ǧ���������п������༰����������ʣ��о�����ռ�ṹ����3�ֵ��������ܴ��ṹ�͵������ʵ�Ӱ�졣�������������ṹ��ԥ���·�Ǧ��(100)�����϶�������ӱ�������ӻ�Ծ����������(100)�����϶��խ��������ʾ��һ���Ľ����ԡ���3�������ԭ��Mulliken��ɵķ�����������п��(110)����ͷ�Ǧ��(100)����ĵ��Ӵ�����������ת�ƣ���������(100)����ĵ�����ӱ���������ת�ơ��Ի����������(100)��(210)��(110)������в�ͬ��λ������ԭ�ӵ�̬�ܶȷ�����������ԭ����λ���ļ��٣�����Fe 3d�����ܼ����ߣ�����̬�ܼ����
�ؼ��ʣ��ռ�ṹ���ܶȷ������ۣ��������ʣ��ܴ��ṹ
��ͼ����ţ�TD923.13���� ���ױ�־�룺A
����������ڼ��Ķ��ѣ�����ԭ���������ڲ�ƽ��״̬������ԭ��֮��ľ�����Ҫ���µ������Դﵽ�µ�ƽ�⣬���������Ϊ�����ԥ�����ڱ���ԭ����λ���ı仯���Լ�ԭ�Ӽ����ĵ��������ӷֲ�Ҳ�������Ӧ���������⣬���ڱ���ԭ�Ӳ��ٴ����������Ƴ������ã�������ܼ�Ҳ�ᷢ���仯��������ӻ������ͬ��������ӵ����ʡ��羧��������ɱ���ʱ����϶�в����ĵ���Tamm����̬[1]���۾����������Ҽ�����϶�в�����Schockley����̬[2]��
����ĸ�ѡ��һ���绯ѧ���̣��о�����խ����������������ı����ڽ�ЧӦ(Surface proximity effect)[3]���������ṹӰ����淴Ӧԭ�ӵĵ������ʣ��Ӷ�Ӱ������ĸ�ѡ�绯ѧ��Ϊ�����⣬�������ԭ���Ǹ�ѡҩ�����ӷ����������õĵط����������ṹ��ѡҩ��������Ҫ�ڽṹ���������ƥ�䣬��ѡҩ�������ڿ�������Ϸ�����Ч����[4-5]����ˣ��о��������ռ�ṹ��������ܴ��ṹ�͵������ʵ�Ӱ�������Ҫ�����������ʵ����ֵ��
Ŀǰ���ܶ�ѧ�߲���X���߹��������(XPS)[6-13]��ɨ�������羵(STM)[14-17]��������������(UPS)[18-20]�����ܵ�������(LEED)[21-23]�ͳ��������Ѹ���(TPD)[24-25]�Ȳ��Է����о���Ǧ���������п��ı������ʡ����⣬Ҳ���о��߲���Hartree-Fock[26-29]���ܶȷ�������[30-38]����3������Ĵ�ģ�ͻ�������ģ�ͽ����о����㡣��Ȼ������������ʵ�ʵ���о���ģ���о��Ѿ��кܶ࣬���Ǵӱ���ռ�ṹ�����������������������ʵ��о���δ����������
�������߲����ܶȷ������ۣ������˷�Ǧ���������п������������ģ�ͣ��о�����3�ֵ����������ռ�ṹ���ܴ��ṹ�͵������ʵ�Ӱ�죬�����˱���ԭ����λ�뻯ѧ���ԵĹ�ϵ���о���������︡ѡ�绯ѧ�����о���ҩ��������ƾ��вο���ֵ��
1 ���㷽����ģ��
���о��ļ�����û����ܶȷ������۵�Materials Studio ���������е�CASTEPģ�����[39]����Ǧ��ͻ���������н���������������GGA-PW91����п��ļ������GGA-PBE [40-41]�����ó�������(Ultrosoft)��������ʵ�ͼ۵��ӵ������[42]����ԭ�ӵ����Ƽ���ѡȡ�ļ۵��ӷֱ�ΪPb 5d106s26p2��Fe 3d64s2��Zn 3d104s2��S 3s23p4�����Ż�����ԭ����ϵ�ļ��ι����п�������������������ƽ�沨�ض��ܵIJ��Խ������Ǧ���������п�������������õĽض��ֱܷ�Ϊ270 eV��280 eV ��310 eV�� Brillouin���Ļ��ּ������Monkhorst-Pack����[43]��ѡ��k�����ֱ�Ϊ1��2��1��2��2��1��2��3��1���Ա�֤��ϵ�����������걸ƽ�沨��ˮƽ�ϵ�����������Ǣ�������У�������Pulay�ܶȻ�Ϸ�����Ǣ������������Ϊ2.0��10-6 eV/atom���ڶ�ģ�͵Ľṹ�Ż��в���BFGS�㷨���Ż���������ԭ�Ӽ������������������Ϊ0.05 eV/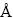 ��������Ӧ������������Ϊ0.1 GPa��ԭ�����λ����������Ϊ2��10-3 ������3������ͬʱ�����Ż����ṹ�Ż���ɵı�־����Щ�������ﵽ���������������������⡣
��������Ӧ������������Ϊ0.1 GPa��ԭ�����λ����������Ϊ2��10-3 ������3������ͬʱ�����Ż����ṹ�Ż���ɵı�־����Щ�������ﵽ���������������������⡣
ѡȡ�˷�Ǧ��(100)�棬������(100)�����п��(110)����Ϊ�о�������3�ֱ����Ϊ�ȶ�������[44]����ԭ�Ӳ�������ղ��Ȳ��Ժ��ڷ�Ǧ��(100)���ȡ 8 ��ԭ�Ӳ㣬������(100)���ȡ15��ԭ�Ӳ㣬��п��(110)���ȡ10��ԭ�Ӳ㣬��ղ��Ⱦ�Ϊ 15 ��3���������ģ����ͼ1��ʾ��
2 ���������
2.1 �����ܴ��ṹ
���ڿ���������ά�����Խṹ��ɶ�ά�ṹ��������������ԭ�ӵIJ������ᷢ���������Ӷ����±���̬������֮���������ã�ʹ�����ܼ�չ��Ϊ�����ܴ�����ͬ�Ŀ������ṹ��Ӱ��ԭ�Ӽ�IJ��������ã��Ӷ��γɲ�ͬ��������ܴ��ṹ�������Ծ��в�״�ṹ�ķ�Ǧ��;�������ռ�ṹ�Ļ�����Ϊ���������ۿռ�ṹ�Ա����ܴ��ṹ��Ӱ�졣
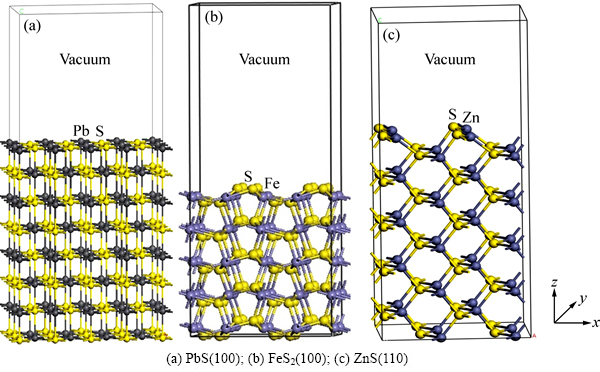
��Ǧ�������(100)�����ܴ��ṹ��ͼ2��ʾ����ͼ2�ɼ�����Ǧ������ܴ���������ܣ��������ڲ��õı���ģ��ԭ������������Ե�ʡ��ȽϷ�Ǧ������ͱ�����ܴ��ṹ����Ǧ�����Ľ������ȴ�����0.5 eV����0.7 eV��˵����Ǧ�������ӽṹ�����������仯�����ӴӼ۴�ԾǨ��������Ҫ��������������ⷽǦ������ܴ��ṹҲ���������Ա仯��1) ��Ǧ����浼�����ܴ���������飬��������������һ���ܴ���˵������ij��ֵ��µ����ܼ��������ѣ����������ܼ�����һ���ܴ�������õ��ӣ�2) ��Ǧ�����۴������Ա����Ǧ������������ܼ������շ��ӹ�����ۣ��۴��������ǵ��������������ӵ��ܼ�����˷�Ǧ�����ĵ��ӱ����������ʧȥ���ӡ�
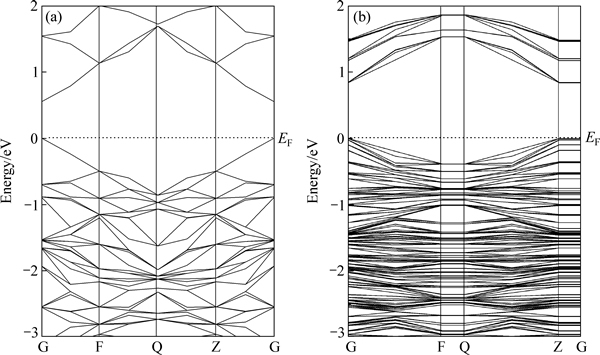
ͼ3��ʾΪ�����������(100)�����ܴ��ṹ�����ڻ�������棬���ܴ��ṹ�����������Ե��������Ȼ��������������ȱ�խ��ֻ��0.45 eV�����������Ϊ0.62 eV����һ����ͷ�Ǧ��������෴�ģ���Ǧ�����Ľ������ȱ�����Ĵ�˵��������ͷ�Ǧ���ڰ뵼�������Ͼ������ԵIJ�ͬ����Σ����������۴����ƣ�˵��������õ���������ǿ�������״����ܴ��߱�����ƽ����˵����������浼��������Ч���������ӵľ����Ա�ǿ��
���������ۿ�֪����Ǧ�����ൽ������һ���������ȱ��Ĺ��̣������������ൽ������һ���������ȱ�С�Ĺ��̡�ͼ4��ʾΪ��Ǧ��ͻ��������ֿ����ڱ����ܴ��ı仯ģ�͡�
ͼ1 �������㾧ģ��
Fig. 1 Slab models of sulfide mineral surface
ͼ2 ��Ǧ�������(100)�����ܴ��ṹ
Fig. 2 Band structure of bulk galena (a) and (100) surface (b)
ͼ3 �����������(100)�����ܴ��ṹ
Fig. 3 Band structure of bulk pyrite (a) and (100) surface (b)
���������ǰ뵼���һ����Ҫ�������������С�뾧��ṹ��ԭ�ӵĽ�����ʵ��йء��������ȵĴ�Сʵ�����Ƿ�ӳ�˼۵��ӱ�����ǿ���̶ȵ�һ����������Ҳ���Dz���������������Ҫ����С����������ǰ��Է�Ǧ��ͻ��������ṹ�ij�ԥ�о�[45-46]���֣���Ǧ�������нϴ�ij�ԥ����������������ԥ��С����ˣ������Ʋ������ͷ�Ǧ�������������������ȵı仯���ɲ�ͬ��ԭ����ɵġ����������������ȵļ�С�����ɱ����ԥ��ɵģ��������ڱ���ԭ����λ����С���Լ۵������������������Ӷ���С�˼۵���ԾǨ������������������Ǧ�����ԭ�ӵ���λ����Ȼ���������Ҳ�Ǽ�С�ģ���������Ǧԭ�ӵ�ԭ�������ϴԼ۵����������ý�ǿ���Ӷ���������λ���仯��Ӱ�죬��Ǧ�����ṹ�ϴ��ԥ�Լ۵��ӵ�����������������Ӱ�죬���µ���ԾǨ��������
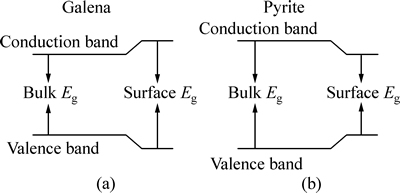
ͼ4 ��Ǧ��ͻ���������ܴ�ģ��
Fig. 4 Surface band structure model for galena (100) (a) and pyrite (100) (b)
�ӱ���������ȱ仯�������������ڷ�Ǧ�����Լ۵��ӵ������̶�Ҫ�Ȼ���������ǿ����ˣ�������Ϊ��Ǧ�����ĵ��ӻ���Ҫ�Ȼ�������������о����֤ʵ���ϻ��ಶ�ռ��ڷ�Ǧ����涼���γɽ����Σ�û�е绯ѧ�����IJ��ռ��������ҩ����ҩ�����Ȳ��ռ��ڻ�������涼���Է����绯ѧ���������γɲ��ռ������
2.2 ����Mulliken��ɷֲ�
����ṹ������ƽ��Ĺ����У��������νṹ�ᷢ���ع�������ԭ���ϵĵ��Ҳ�ᷢ�����·ֲ�����1~3����Ϊ��п��(110)�桢������(100)��ͷ�Ǧ��(100)�����ԭ�ӵ�ɷֲ������
��п��(110)����ԭ�ӽṹ�ͱ����ͼ5��ʾ���ӱ�1�ɼ�������п����漸���ع������У��������ԭ�ӵ�ɷ������·ֲ���пԭ�ӵ�ɷֲ��仯�ϴ���ԭ�ӱ仯��С����п����ṹ��1��пԭ�Ӻ�4����ԭ����λ��1����ԭ�Ӻ�4��пԭ����λ��п����ԭ��Ϊ����λ�ṹ�����ڱ���ԭ�Ӽ��Ķ��ѣ�������ԭ�������ֽṹ��������λ��Zn1��S1������λ��Zn2��S2����ͬ��λ����ԭ�ӺͲ�ͬλ�õ�ԭ�ӵĵ���ת�ƺͷֲ���ͬ��
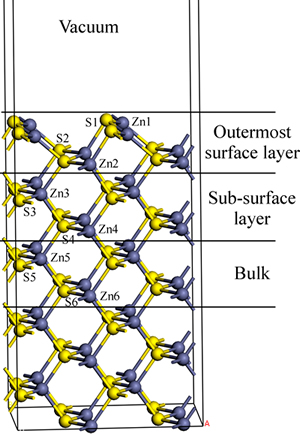
ͼ5 ��п��(110)����ṹ��ԭ��λ��ʾ��ͼ
Fig. 5 Schematic diagram of sphalerite (110) surface structure
��ԭ�Ӽ۵��ӹ���Ϊ3s23p4��пԭ�Ӽ۵��ӹ���Ϊ3d104s24p0����п���������ԭ��3s���ʧȥ���ӣ�3p����õ����ӣ�пԭ��4s��4p��������ӻ����ã�ͬʱ����Ϊʧȥ���ӣ�3d������ڴ��ڽ����ܼ���ͬʱΪ�������ȶ��ṹ���������������ã���ˣ��������ԭ�Ӻ�пԭ�ӵ���ת����Ҫ����s�����p��������ڴα�����ԭ�ӣ���Ȼ������һ������������λ�ṹ�������������ӵ������㣬��z�����в��Գƽṹ����ͬλ����ԭ�ӵ�ɱ仯������ͬλ�õ�пԭ��ȴ�нϴ�IJ��죬Zn3��Zn4ԭ�ӵ�3d��4p�������û�з����仯��3s���ȴ�нϴ���졣��������пԭ�ӣ��α����Zn4ԭ��4sʧȥ���ӣ�Zn3ԭ��4sȴ�õ����ӣ����ֳ��ռ�ṹ��пԭ�ӵõ���������Ӱ��(п4s���Ϊ��Ծ�������)�������ṹ��пԭ�Ӻ���ԭ�ӣ���λ��Zn1��S1���ڼ��Ķ��ѣ�ֻ������λ����λ��Zn2��S2��Ȼ������λ�������Ը�λ��ԭ�Ӻ�пԭ��������λ���ļ��ٻ�������ӷֲ��Ľϴ�ı䡣��λ��ԭ��(S1)3s�������˽϶�ĵ��ӣ�S1ԭ�ӵĵ��Ҳ�������Ҫ������λ��ԭ��(S2)������λ��û�з����仯��ֻ�ǿռ�ṹ�ĶԳ��Է����仯������ڵ����ֻ�н�С�仯�������ӽ������ɡ�����пԭ�ӣ���λпԭ�Ӻ͵�λпԭ�ӵĵ�ɶ������˽ϴ�仯��������пԭ����ȣ�пԭ�ӵ�4s��������������ӣ�4p�����ͬ��ֻ�и�λпԭ��(Zn1)4p���ʧȥ���ӣ���λпԭ��(Zn2)û�����仯��˵����λ����λ��пԭ�ӵ��ӷֲ���Ӱ�����
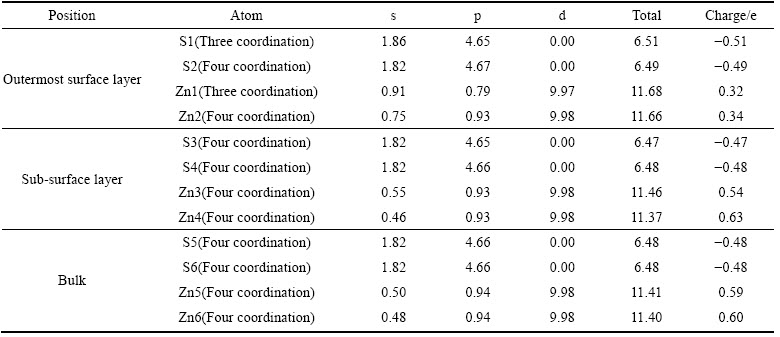
��1 ��п��(110)�����ԥ��ԭ�ӵ�Mulliken��ɷֲ�
Table 1 Mulliken charge of sphalerite (110) surface after relaxation
�����Ϸ������Կ�������п��������ԭ�Ӷ�����������˽϶�ĵ��ӣ�˵����п����淢���˵��Ӹ�������������λпԭ��4s����������������ӣ�����������λпԭ�ӵ����ԣ��������ϻ��มѡ���ռ����ӵ����á�
ͼ6 ��������治ͬ��ԭ�ӱ��
Fig. 6 Schematic plots of Mulliken charge of pyrite (100) surface
��2 ����������ԥ��ԭ�ӵ�Mulliken��ɷֲ�
Table 2 Mulliken charge of pyrite (100) surface after relaxation
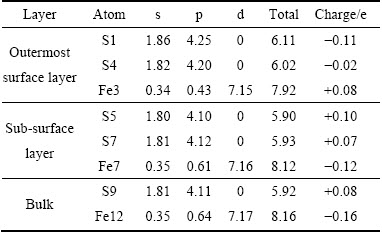
������(100)����ԭ��λ����ͼ6��ʾ���ɱ�2�ɼ��������ൽ����㣬��������ԭ�Ӻ���ԭ�ӵ����һ���dz����Եı仯���Ǿ�����ԭ�Ӵ����������ɱ䵽����ĸ���ɣ���ԭ�Ӵ����ฺ��ɱ䵽���������ɡ�������㵽�����㣬��ԭ�ӵ�3s��3p������������ӣ�����3p����ϵĵ����������ӣ�˵���������ԭ���ڵ������ƽ������л������ԭ�ӵĵ��ӡ�������ԭ�ӣ�������㵽�����㣬����4s��3d����ϵ������������䣬4p����ϵ����������Լ��٣�������0.64���ٵ�0.43��˵���������ԭ�ӵ�4p���ʧȥ�˵��ӣ������������ԭ��3p����õ������Ӧ��˵���ڱ���ԭ���ع��͵������ƽ������У���ɴ���ԭ����ת�Ƶ�����ԭ���ϣ���������ԭ�Ӻ�ԭ�ӵĵ���ת�Ʒ��������������p�������һ������������������ԥ��С�йء�XPS[30]���Խ��Ҳ���������������������ԭ������ԭ��Ǩ�ơ�
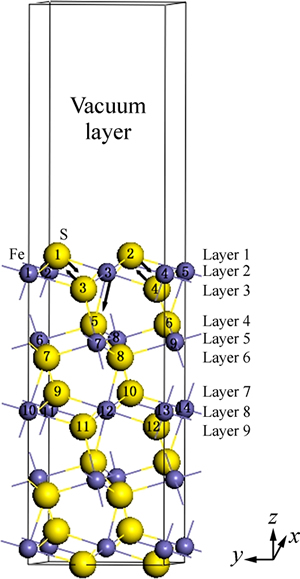
һ����˵�������ķ����ܼ����ڰ뵼�����Ի��������̬�ܶȵ��о����[46]����������(100)�������һЩ�ƽ�������������Ϊ�뵼�壬��ˣ������дӱ���������������ƣ����������������������������������ԭ�ӵ�ɵķ���֤ʵ����һ�������ͼ7��ʾ��ÿ6��ԭ�Ӳ�������ԭ������һ������ˣ�6��ԭ�Ӳ����1�����ڣ����дӱ��濪ʼ��һ����ԭ�Ӳ�Ϊ����㣬�ڶ�����ԭ�Ӳ���Կ�������㣬ͼ7�е�����Ϊ������ԭ�Ӳ�ԭ�ӵ��ܵ�ɡ��������������������ĵ��Ϊ-0.33 e��������������Ϊ-0.97 e����˵������㴦�ڵ��Ӹ���״̬������㴦�ڵ���ȱʧ״̬����ˣ�������(100)�����������յ��ӵ��������ڸ�ѡʵ���У����ڻ������������������ԣ���ҩ�ϵĵ�����������������ת�ƣ��Ӷ�����������Ӧ���γ�˫��ҩ��
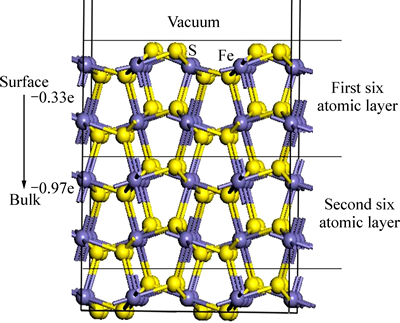
ͼ7 ������(100)������ɱ仯ʾ��ͼ
Fig. 7 Schematic plots of Mulliken charge of pyrite (100) surface
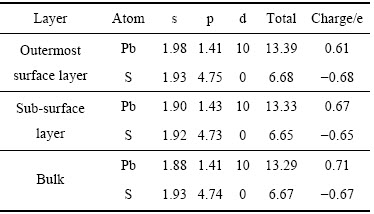
��Ǧ�������в�״�ṹ��ÿһ���Ǧԭ�Ӻ���ԭ�ӵ�λ�ö�������ͬz���꣬����ṹ��Լ������ԥ��ķ�Ǧ�����3���Mulliken������3���С��ɱ�3�ɼ�����ԭ�ӵ�ɴ�����㵽�������-0.67 e�䵽-0.68 e���仯������Ǧԭ�ӵ��ȴ�������ı仯����������0.71 e���ٵ��α�����0.67 e���ټ��ٵ�������0.61 e����Ǧ�����Ǧԭ�ӵ���д����ൽ����ݼ������ơ��ӵ��ӵĹ���ֲ�������Ǧԭ�Ӽ۵��Ӳ�Ϊ 5d106s26p2����ԭ�Ӽ۵��Ӳ�Ϊ3s23p4����Ǧ��������Ҫ����3p�����Ǧ6p����������ã�Ǧ6p����ϵĵ���ת�Ƶ���3p����ϡ��ڷ�Ǧ�����ṹ��ԥ�����У���3p�����Ǧ6p����ϵĵ��ӻ���û�з����仯��˵����Ǧ�����ṹ��ԥ�����ϲ��ı���3p�����Ǧ6p��������ã�����������Ǧԭ��6s����ϵĵ���ȴ�������࣬��������6s1.88����������6s1.98���ӷ�Ǧ�������ӷֲ��͵�ɱ仯��������Ǧ��������ܵ������������Ҫ����ɵı仯���Ǵ������0.04 e�仯���������-0.07 e��˵����Ǧ�������и������ӵ����ʡ���Ǧ�������һ���ʲ����ڻ�ҩ���������γ�˫��ҩ���о������������Ǧ�����Ŀǰֻ����ԭ��Ǧ��û�з���˫��ҩ��
��3 ��Ǧ��(100)�����ԥ��ԭ�ӵ�Mulliken��ɷֲ�
Table 3 Mulliken charge of galena (100) surface after relaxation
2.3 �������̬�ܶ�
�������Ͻ���ԭ����λ��Խ�٣��Լ۵�������ԽС�����ӵľ����Լ�������������ǿ��ԭ�ӷ�Ӧ����Խǿ�� ���ڱ���ṹ�IJ��Գ��ԣ����ű���ṹ�ij�ԥ���������Ҳ�ᷢ�����·ֲ����γɱ������̬��TAMM[1]����1932�����������ھ������������������϶�б���̬�ܼ������ھ���d���ӵı�����˵(�����ﶼ����d����)�����������ԭ�ӵ���λ��Ҫ��������ԭ�ӵ���λ���٣��Ӷ�����d���ӵ�����������ʹԭ���ȽϾ����d̬�����˸߳�3d���ܴ���d���ӱ����ܼ���������Tamm̬��
ͼ8��ʾΪ����������ͱ�����в�ͬ��λ������ԭ�ӵ���̬�ܶȣ�����������ԭ�ӿ��Կ�����λ��Ϊ0�������������ͼ8�ɼ���������λ��Ϊ0��������ԭ�ӣ�������ܼ����ĵ���̬�ܶ���Ҫ����3d��4s���ɣ�����ԭ����λ����3���ӵ�6��ʱ�����ܼ�����p�����s�������̬��ʧ��ֻʣ��3d̬��˵��������ԭ�ӵ���λ���ã���ǿ�˶���ԭ�ӵ��ӵ������̶ȡ��������ԭ��3d����̬�ı仯������ԣ���ͬ�ṹ�µ���ԭ��3d̬���кܴ�IJ�ͬ������(110)������λ��ԭ�ӵ�3d����̬��������ǿ�����û�з������ѣ���������λ������ԭ�ӣ�������(210)���������λ��������(100)���������λ����(110)���������λ������3d��������Է���Ϊ3���֣����з����ܼ���Ϊt2g�Ǽ������������������eg�ɼ������eg*�������������ԭ�ӱ������̬������������ԭ�ӵĵ���̬�ܶ�������λ���ļ��٣���3d̬Խ��Խ�����ܼ������ƶ�������λ���ļ��٣�����3d�����ܼ����ߣ��γɱ���̬�ܼ���
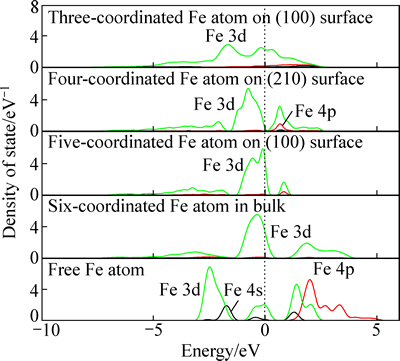
ͼ8 ������ͬ��λ������ԭ�ӵĵ���̬�ܶ�
Fig. 8 DOS of pyrite Fe atom with different coordination number
3 ����
1) ����ṹ��ԥ�ᵼ�¿�������ܴ��ṹ�����仯����Ǧ��(100)����������ȱ��������(100)��������������խ���������һ���Ľ����ԣ������ڱ�����ԭ�ӵ����������нϲ�����ռ���ѡ��Ϊ��
2) ���滯ѧ���Ķ��ѶԱ���ԭ�ӵ�ɷֲ�����Ӱ�졣��п��(110)���淢���˵��Ӹ�������������λпԭ��4s����������������ӣ�����������λпԭ�ӵ����ԣ������ڻ�ҩ�ಶ�ռ������á�������(100)����������������������Ӵӱ���������ת�ƣ�ͬʱ����������沿�ֵ�ɴ���ԭ��ת�Ƶ���ԭ���ϣ������ڻ�ҩ����Ϊ˫��ҩ��Ӧ�Ľ��С����ڷ�Ǧ����(100)�����ܵ�ɱ�����࣬������Ӹ����������ڻ�ҩ�ĵ绯ѧ�������ã��ڷ�Ǧ����������ڲ��ռ����ӷ�����ѧ�������á�
3) ����ԭ����λ���ĸı�ᵼ�¿���������̬���γɡ������������(100)��(210)��(110)������ԭ�ӵ�̬�ܶȽ��������������ԭ����λ�����٣�����Fe 3d�����ܼ����ߣ�����̬�ܼ��������������λ����ԭ���Ǹ�ѡ�����з������õı�����������λ��
REFERENCES
[1] Tamm I E. On the possible bound states of electrons on a crystal surface[J]. Physcs Z Soviet, 1932, 1: 733-746.
[2] Shockley W. On the surface states associated with a periodic potential[J]. Physical Review, 1939, 56: 317-323.
[3] Becker U, Rosso k M, Hochella M F J R. The proximity effect on semiconducting mineral surfaces: A new aspect of mineral surface reactivity and surface complexation theory[J]. Geochimica et Cosmochimica Acta, 2001, 65(16): 2641-2649.
[4] RAI B. Molecular modeling and rational design of flotation reagents[J]. International Journal of Mineral Processing, 2003, 72(1/4): 95-110.
[5] �� ��, �� ��, �� ˧, �� Ԫ. ���ռ����о���չ[J]. Ӧ�û���, 2012, 41(10): 1791-1795.
MA Xin, ZHONG Hong, WANG Shuai, HU Yuan. Research on the sulfide ore collectors[J]. Applied Chemical Industry, 2012, 41(10): 1791-1795.
[6] Heide H V, Hemmel R, Bruggen C F V, Haas C. X-ray photoelectron spectra of 3d transition metal pyrites[J]. Journal of Solid State Chemistry, 1980, 33(1): 17-25.
[7] Chernyshova I V, Andreev S I. Spectroscopic study of galena surface oxidation in aqueous solutions I. Identification of surface species by XPS and ATR/FTIR spectroscopy[J]. Applied Surface Science, 1997, 108: 235-236.
[8] Leiro J A, Mattila S S, Laajalehto K. XPS study of the sulphur 2p spectra of pyrite [J]. Surface Science, 2003, 547: 157-161.
[9] Cai Yuan-feng, Pan Yu-guan, Xue Ji-yue, Sun Qing-feng, Su Gui-zhen, Li Xiang. Comparative XPS study between experimentally and naturally weathered pyrites[J]. Applied Surface Science, 2009, 255: 8750-8760.
[10] Laajalehto K, Kartio I, Suoninen E. XPS and SR-XPS techniques applied to sulphide mineral surfaces[J]. International Journal of Mineral Processing, 1997, 51:163-170.
[11] Mycroft J R, Bancroft G M, McIntyre N S, Lorimer J W, Hill I R. Detection of sulphur and polysulphides on electrochemically oxidized pyrite surfaces by X-ray photoelectron spectroscopy and Raman spectroscopy[J]. Journal of Electroanalytical Chemistry and Interfacial Electrochemistry Electrochem, 1990, 292: 139-152.
[12] Nowak P, Laajalehto K. Oxidation of galena surface-An XPS study of the formation of sulfoxy species[J]. Applied Surface Science, 2000, 157: 101-111.
[13] Harmer S L, Goncharova L V, Kolarova R, Lennard W N,  M A, MITCHELL I V, Nesbitt H W. Surface structure of sphalerite studied by medium energy ion scattering and XPS[J]. Surface Science, 2007, 601: 352-361.
M A, MITCHELL I V, Nesbitt H W. Surface structure of sphalerite studied by medium energy ion scattering and XPS[J]. Surface Science, 2007, 601: 352-361.
[14] Becker U, Michael F, Hckhella J R. The calculation of STM images, STS spectra, and XPS peak shifts for galena: New tools for understanding mineral surface chemistry[J]. Geochimica et Cosmochimica Acta, 1996, 60: 2413-2426.
[15] Laajalehto K, Smart R St C, Ralston J, Suoninen E. STM and XPS investigation of reaction of galena in air[J]. Applied Surface Science, 1993, 64: 29-39.
[16] Eggleston C M, Hochella M F. Scanning tunneling microscopy of pyrite {100} surface structure and step reconstruction[J]. American Mineralogist, 1992, 77: 221-224.
[17] Mikhlin Y L, Romanchenko A S. Gold deposition on pyrite and the common sulfide minerals: An STM/STS and SR-XPS study of surface reactions and Au nanoparticles[J]. Geochimica et Cosmochimica Acta, 2007, 71(24): 5985-6001.
[18] Sugaa S, Tabiguchia M, Shina S, Sekia M, Shibuya S, Sato K, Yamaguchi T. Reflectance and UPS studies of band structures and final state interactions of low-spin pyrites[J]. Physica B+C, 1983, 117/118: 353-355.
[19] Ballester A, Bl��zquez M L, Gonz��lez F, Rom��n E, Bustillo F J. Studies of zinc sulphide, treated with different solutions of catalyst ions[J]. Vacuum, 39(7/8): 663-664.
[20] Muscata J, Klauber C A. Combined ab initio and photoelectron study of galena (PbS)[J]. Surface Science, 2001, 491: 226-238.
[21] Chaturvedi S, Katz R, Guevremont J, Schoonen M A A, Strongin D R. XPS and LEED study of a single-crystal surface of pyrite[J]. American Mineralogist, 1996, 81: 261-264.
[22] Rosso K M, Becker U, Hochella J R M F. Atomically resolved electronic structure of pyrite {100} surfaces: An experimental and theoretical investigation with implications for reactivity[J]. American Mineralogist, 1999, 84: 1535-1548.
[23] Duke C B, Wang Y R. Surface structure and bonding of the cleavage faces of tetrahedrally coordinated II�CVI compounds[J]. Journal of Vacuum Science Technology B, 1988, 6: 1440-1443.
[24] Guevremont J M, Strongin D R, Schoonen M A A. Effects of surface imperfections on the binding of CH3OH and H2O on FeS2(100): Using adsorbed Xe as a probe of mineral surface structure[J]. Surface Science, 1997, 391: 109-124.
[25] Guevremont J M, Strongin DR, Schoonen M A A. Thermal chemistry of H2S and H2O on the (100) plane of pyrite: Unique reactivity of defect sites[J]. American Mineralogist, 1998, 83: 1246-1255.
[26] Becker U, Greatbanks S P, Rosso K M, Hillier I H, Vaughan D J. An embedding approach for the calculation of stm images: Method development and application to galena (PbS)[J]. The Journal of Chemical Physics, 1997, 107: 7537-7542.
[27] Mian M, Harrison N M, Saunders V R, Flavell W R. An ab initio Hartree-Fock investigation of galena (PbS)[J]. Chemical Physics Letters, 1996, 257: 627-632.
[28] Wilson N, Muscat J. The calculation of structural, elastic and phase stability properties of minerals using first principles techniques: A comparison of HF, DFT and hybrid functional treatments of exchange and correlation[J]. Molecular Simulation, 2002, 28: 903-915.
[29] Steele H M, Wright K, Hillier I H. A quantum-mechanical study of the (110) surface of sphalerite (ZnS) and its interaction with Pb2+ species[J]. Physics and Chemistry of Minerals, 2003, 30(2): 69-75.
[30] von Oertzen G U, Skinner W M, Nesbitt H W. Ab Initio and XPS studies of pyrite (100) surface states[J]. Radiation Physics and Chemistry, 2006, 75(11): 1855-1860.
[31] CHEN Jian-hua, Lan Li-hong, CHEN Ye. Computational simulation of adsorption and thermodynamic study of xanthate, dithiophosphate and dithiocarbamate on galena and pyrite surfaces[J]. Minerals Engineering, 2013, 46/47: 136-143.
[32] Stirling A, Bernasconi M, Parrinello M. Defective pyrite (100) surface: An ab initio study[J]. Physical Review B, 2007, 75: 165406 1-8.
[33] CHEN Jian-hua, CHEN Ye, LI Yu-qiong. Effect of vacancy defects on electronic properties and activation of sphalerite (110) surface by first-principles[J]. Transactions of Nonferrous Metals Society of China, 2010, 20(3): 502-506.
[34] Hung A, Muscat J, Yarovsky I, Russo S P. Density-functional theory studies of pyrite FeS2 (100) and (110) surfaces[J]. Surface Science, 2002, 513: 511-524.
[35] CHEN Jian-hua, WANG Lei, CHEN Ye, GUO Jin. A DFT study of the effect of natural impurities on the electronic structure of galena[J]. International Journal of Mineral Processing, 2011, 98: 132-136.
[36] Muscat J, Gale J D. First principles studies of the surface of galena PbS[J]. Geochimica et Cosmochimica Acta, 2003, 67: 799-805.
[37] Hung A, Muscat J, Yarovsky I, Russo S P. Density-functional theory studies of pyrite FeS2 (111) and (210) surfaces[J]. Surface Science, 2002, 520: 111-119.
[38] �½���, ��С��, �� ��, �Ż���. ����λ������ȱ�ݵ���п����ӽṹ�ĵ�һ��ԭ������[J]. �й���ɫ����ѧ��, 2010, 20(4): 765-771.
CHEN Jian-hua, ZENG Xiao-qin, CHEN Ye, ZHANG Hui-peng. First-principle theory calculations of electronic structure of sphalerite with vacancy and impurity[J]. The Chinese Journal of Nonferrous Metals, 2010, 20(4): 765-771.
[39] Payne M C, Teter M P, Allan D C, Arias T A, Joannopoulos J D. Iterative minimization techniques for ab initio total energy calculation: Molecular dynamics and conjugate gradients[J]. Reviews of Modern Physics, 1992, 64: 1045-1097.
[40] PERDEW J P, BURKE K, ERNEZERHOF M. Generalized gradient approximation made simple[J]. Physical Review Letter, 1996, 77: 3865-3868.
[41] Perdew J P, Wang Y. Accurate and simple analytic representation of the electron-gas correlation energy[J]. Physical Review B, 1992, 45: 13244-13249.
[42] Vanderbilt D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism[J]. Physical Review B, 1990, 41: 7892-7895.
[43] Monkhorst H J, PACK J D. Special points for Brillouin-zone integrations[J]. Physical Review B, 1976, 13: 5188-5192.
[44] Vaughan D J, Becker U, Wright K. Sulphide mineral surfaces: Theory and experiment[J]. International Journal of Mineral Processing, 1997, 51(1/4): 1-14.
[45] ������, �½���, ������, �� ��. �������ڻ�����ͷ�Ǧ����������[J]. �й���ɫ����ѧ��, 2012, 22(4): 1184-1194.
LI Yu-qiong, CHEN Jian-hua, LAN Li-hong, GUO Jin. Adsorption of O2 on pyrite and galena surfaces[J]. The Chinese Journal of Nonferrous Metals, 2012, 22(4): 1184-1194.
[46] ������, �½���, �� ��, �� ��. ������(100)�������ʵ��ܶȷ������ۼ��㼰��Ը�ѡ��Ӱ��[J]. �й���ɫ����ѧ��, 2011, 21(4): 919-926.
LI Yu-qiong, CHEN Jian-hua, CHEN Ye, GUO Jin. Density functional theory calculation of surface properties of pyrite (100) with implications for flotation[J]. The Chinese Journal of Nonferrous Metals, 2011, 21(4): 919-926.
Effect of spatial structure on band structure and electronic properties of sulphide minerals
CHEN Ye1, 2, CHEN Jian-hua1, 2, LI Yu-qiong1, 2, ZHAO Cui-hua3
(1. College of Resources and Metallurgy, Guangxi University, Nanning 530004, China;
2. Guangxi Colleges and Universities Key Laboratory of Minerals Engineering, Guangxi University, Nanning 530004, China;
3. School of Materials Science and Engineering, Guangxi University, Nanning 530004, China)
Abstract: The bulk and surface electronic properties of galena, pyrite and sphalerite were calculated by adopting density functional theory, and the effects of spatial structure on the band structure and electronic properties of these three typical sulphide minerals were studied. The results show that surface relaxation leads a greater band gap of PbS (100) surface compared with the bulk PbS, and the surface electrons are more reactive than the bulk electrons, while for the FeS2 (100) surface, the band gap decreases and shows a metallic characteristics. The analysis of surface Mulliken charge of these three sulphide minerals suggests that, for the PbS (100) and ZnS (110), the electrons transfer from the bulk to the surface, however, the electrons of FeS2 (100) transfer from the surface to the bulk. The DOS of bulk pyrite and surface pyrite Fe atom with different coordination number indicate that the decrease of coordination number leads to the increase of Fe 3d energy level and Tamm surface energy level.
Key words: sulfide mineral; spatial structure; density functional theory; electronic property; band structure
Foundation item: Projects(51574092, 51364002, 51304054) supported by the National Natural Science Foundation of China; Project(2014GXNSFAA118316) supported by Gangxi Natural Science Foundation, China
Received date: 2015-10-19; Accepted date: 2016-03-18
Corresponding author: CHEN Jian-hua; Tel: +86-771-3232200; E-mail: jhchen@gxu.edu.cn
(�༭ �� ��)
������Ŀ��������Ȼ��ѧ����������Ŀ(51574092��51364002��51304054)��������Ȼ��ѧ����������Ŀ(2014GXNSFAA118316)
�ո����ڣ�2015-10-19�������ڣ�2016-03-18
ͨ�����ߣ��½��������ڣ���ʿ���绰��0771-3232200��E-mail��jhchen@gxu.edu.cn

